分子レベルの構造生物学の推進に向けて
「第4部報告」
平成12年6月26日
日本学術会議第4部
この報告は、第17期日本学術会議第4部附置分子レベルの構造生物学振興推進小委員会で審議した結果を取りまとめ、第4部報告として発表するものである。
第4部附置分子レベルの構造生物学振興推進小委員会
委員長 郷 信広 日本学術会議第4部会員,京都大学大学院理学研究科教授
幹事 片岡 幹雄 奈良先端科学技術大学院大学教授
〃 中村 春木 大阪大学蛋白質研究所教授
〃 難波 啓一 ERATO代表研究者
委員 植木 龍夫 高輝度光科学研究センター放射光研究所利用促進部門長
〃 内田 久雄 日本学術会議第4部会員,東京大学名誉教授
〃 小笠原直毅 奈良先端科学技術大学院大学教授
〃 甲斐荘正恒 東京都立大学大学院理学研究科教授
〃 倉光 成紀 大阪大学大学院理学研究科教授
〃 黒木 良太 キリンビール(株)医薬探索研究所主任研究員
〃 坂部 知平 高エネルギー物理学研究所名誉教授
国際科学振興財団専任研究員
嶋本 伸雄 国立遺伝学研究所教授
白川 昌宏 奈良先端科学技術大学院大学助教授
田中 勲 北海道大学大学院理学研究科教授
月原 冨武 大阪大学蛋白質研究所教授
藤吉 好則 京都大学大学院理学研究科教授
前田 雄一郎 理化学研究所主任研究員
横山 茂之 東京大学大学院理学系研究科教授理化学研究所主任研究員
日本学術会議第17期第4部会員
部長 和田 昭允 理化学研究所ゲノム科学総合研究センター長
東京大学名誉教授
副部長 大瀧 仁志 立命館大学理工学部応用化学科教授
東京工業大学名誉教授・総合大学院大学名誉教授
幹事 鎮西 清高 大阪学院大学情報学部教授 京都大学名誉教授
幹事 土居 範久 慶應義塾大学理工学部教授
会員 青木謙一郎 東北大学名誉教授
赤岩 英夫 群馬大学学長
荒牧 重雄 日本大学文理学部教授 東京大学名誉教授
池内 了 名古屋大学大学院理学研究科教授
岩槻 邦男 立教大学理学部教授 東京大学名誉教授
上野 健爾 京都大学大学院理学研究科教授
内田 久雄 東京大学名誉教授
江澤 洋 学習院大学理学部教授
岡本 和夫 東京大学大学院数理科学研究科教授
尾本 惠市 国際日本文化研究センター研究部教授 東京大学名誉教授
榧根 勇 愛知大学現代中国学部教授 筑波大学名誉教授
郷 信広 京都大学大学院理学研究科教授
合志 陽一 国立環境研究所副所長
斎藤 常正 東北大学大学院理学研究科教授
坂元 昴 文部省メディア教育開発センター所長
東京工業大学名誉教授・大学入試センター名誉教授
櫻井 英樹 東京理科大学理工学部工業化学科教授 東北大学名誉教授
柴田 徳思 高エネルギー加速器研究機構放射線科学センター長 教授
田中 正之 東北大学理学部大気海洋変動観測研究センター長 教授
戸塚 績 江戸川大学社会学部環境情報学科教授
長岡 洋介 関西大学工学部教授
京都大学名誉教授・名古屋大学名誉教授
廣田 榮治 総合研究大学院大学学長分子科学研究所名誉教授
星 元紀 慶応義塾大学理工学部教授
益川 敏英 京都大学基礎物理学研究所所長教授
森脇 和郎 総合研究大学院大学副学長 国立遺伝学研究所名誉教授
矢原 一郎 財団法人東京都臨床医学総合研究所副所長
吉原經太郎 北陸先端科学技術大学院大学材料科学専攻教授
吉村 功 東京理科大学工学部経営工学科教授
要 旨
[背景]第16期学術会議第4部は「分子レベルの構造生物学振興小委員会」を同部付置で設け、「分子レベルの構造生物学のわが国における振興について」と題する報告書をまとめた。その後もこの研究分野は、激しい研究環境・状況の変化の中にいるため、第17期においても前期の活動を引き継ぎ、現時点での振興・推進策を検討するため、第4部は再び「分子レベルの構造生物学振興・推進小委員会」を同部付置で設けた。本報告書はその小委員会による検討結果を報告書としてまとめたものである。
[問題点]構造生物学は生体高分子およびそれの作る細胞内複合体の立体構造を原子レベルの解像能で明らかにし、それに基づいて生命現象の諸相を明らかにしようとする研究分野である。この分野は、立体構造の実験的解明がいろいろな技術的展開の結果著しく加速されてきたこと、および密接な関係にあるゲノム科学の爆発的展開の結果、現在大きな展開の時期を迎えている。この時点でわが国における構造生物学研究を如何に推進していくべきか、当該分野研究者の深い洞察に基づく判断が必要とされている。小委員会では、この問題を「構造生物学とはなにか」の原点に立ち戻り、議論を重ねて提言をまとめた。
[提言]提言ではこの分野の現状に於ける研究を次の3本柱にまとめ、いずれをも注意深く推進する必要があるとしている。(1)重要な生物学上の問題を構造に基づいて解明する研究。(2)特定の集団の蛋白質の構造を網羅的に調べ上げる研究。(3)最先端の技術・方法を構築する研究。
構造生物学は放射光等の大型施設を用いることを除けば、本質的にはスモールサイエンスであり、さらに、対象・方法の多様性にその特徴がある。この特徴の認識に立脚し、各研究者のキャリアパスを考慮に入れ、国内の構造生物学研究全体の活性を高く保つための多様なサイズ・性格の研究室の全国的配置を骨子とする全国的研究体制を提言している。
第1に、全国5、6箇所程度の拠点に研究複合を設けることを提言し、その具体的な候補も挙げている。これらの拠点に複数の構造解析に携わる研究室と理論研究室からなる研究複合を形成する。
第2に、全国5〜10箇所程度の主要大学に幾つかの研究室からなる研究・教育複合を設立すべきことを提言している。これら拠点大学における研究者育成のための教育面での機能は特に大切である。
第3に、上記以外に全国に100程度の研究室が展開していることが必要である。特に、大学の医学、生物学、農学、薬学等の学部・研究科に構造生物学に携わる研究室を必ず設置することは、構造生物学にとってもわが国のライフサイエンス全体にとっても切実に求められている。
構造生物学は多様な方向に展開しつつ、新たな生命科学の中核を形成しつつある。用いられる方法も多様で、しかも激しく発展しつつある。展開しつつあるいずれの方向においても、純粋な基礎と応用との間の距離が短い。この短さ故、国際的な知的財産権を巡る競争に立ち向かわざるを得ない。この状況の中で、研究のための財政的・物的資源を適切に配分し、人的資源を急速に育成しつつ適切に配分しなければならない。この複雑な問題に研究者社会は叡智を持って立ち向かわなければならない。
![]()
目次
1.構造生物学とはなにか
1.1.構造生物学の定義と現代生物学史における位置付け
1.2.ゲノム科学進展と構造生物学
1.3.構造生物学がもたらすもの
2.立体構造を解明する実験的手段
2.1.X線結晶構造解析
2.2.NMR
2.2.1生体分子の原子レベルでの立体構造決定
2.2.2生体分子間の相互作用の解析
2.2.3蛋白質の運動性の解析
2.2.4蛋白質のフォールディング
2.3.電子顕微鏡
2.3.1.電子顕微鏡による構造研究の特徴
2.3.2.世界および我が国の状況
2.4.中性子散乱・回折
2.4.1.中性子散乱・回折の特徴
2.4.2.近年のわが国と世界の状況
2.4.3.中性子源将来計画
2.5.X線繊維回折・溶液散乱
2.6.走査プローブ顕微鏡、新世代光学顕微鏡
3.立体構造データベース
3.1.世界およびわが国における現状
3.2.バイオインフォーマティクス現状と動向
3.3.立体構造データベース利用法の展開
3.3.1.比較・分類
3.3.2.相互作用の解析
3.3.3.機能予測
4.理論の役割
4.1.特定の系の立体構造情報に基づく機能発現機構の解析
4.2.立体構造の博物学的研究
4.3.立体構造予測法の開発・機能予測
4.4.新しい実験手段開発への寄与
5.構造生物学の主要な課題
5.1重要な生命現象を担うマシナリーの構造決定と機能発現機構の解明
−立体構造と機能理解との間の距離はいろいろ−
5.1.1立体構造が直ちに機能発現の仕組みを示唆する場合
5.1.2多くの条件下での立体構造決定が必要な場合
5.1.3深い理論的解析を必要とする場合
5.2.蛋白質全ファミリーの立体構造決定プロジェクト
5.2.1.意義と可能性
5.2.2.生命科学への波及効果
5.2.3.世界の動きとわが国での対応
5.2.4.問題点
5.3.新しい実験的立体構造決定法の開発
5.3.1.より大きなより複雑な系
5.3.2.揺らぎやダイナミックス情報をも得られる高精度構造決定
5.3.3.時間軸測定
5.3.4.迅速構造解析
5.3.5.構造解析ソフトウエアの開発
5.4.細胞生物学への新しい寄与:空間と時間を扱う広義の構造生物学
5.5.先端医療・工業等への応用
5.5.1.医療への寄与
5.5.2.創薬への寄与
5.5.3.工業への応用
6.わが国の研究・教育体制
6.1.研究体制の現状と問題点
6.1.1.大学などにおける研究体制の現状
6.1.2.共同利用研究施設の現状と問題点
6.1.2.1.SPring-8
6.1.2.2.物質構造科学研究所(放射光実験施設)の現状と問題点
6.1.2.3.NMR共同利用の現状と問題点
6.1.2.4.データベースの現状と問題点
6.2.人材育成体制
7.提言構造生物学推進のあるべき姿
7.1.構造生物学研究の3本柱
7.2.多様なサイズ・性格の研究室の全国的配置
7.3.理論的研究の必要性
7.4.全国的研究体制
7.4.1.拠点研究複合
7.4.2.拠点大学に於ける研究・教育複合
7.4.3.幅広い展開
7.5.構造生物学の総合的展開へ向けた研究者社会の対応
1.構造生物学とはなにか
1.1.構造生物学の定義と現代生物学史における位置付け
「分子生物学(Molecular Biology)」と言う名前は、ペプシンの結晶化に端を発するX線結晶構造解析によって蛋白質の構造を求めようとしていた黎明期に、生体高分子の立体構造情報が生み出すべき新たな生物学の名称として提案された。しかし、分子生物学は、DNAと遺伝子を中心とする新しい生物学の名前となり、生体高分子の構造学は、分子生物学の中心とは独立した進展を、主に生化学の中で遂げてきた。そして、分子生物学が完熟する80年代後半になり、DNAと蛋白質との相互作用を生み出す共通部分構造の探索法等として、構造生物学は再び生物学として復権し始めた。
この歴史は、生体高分子を意識した生物学研究のパラダイムが変化したことを物語っている。黎明期では、蛋白質の原子分解能での構造から生み出される精妙な構造自体驚であり、生物界においても物理や化学の法則が貫徹している信念を確立した。蛋白質が結晶したことから蛋白質は一定の立体構造を持つことが明らかにされ、そのことは今日では「蛋白質は1次構造が決まれば3次構造も決まる」というゲノム科学の中心的ドグマとなっている。
当時の蛋白質構造学は、酵素をその研究の主要な標的としたため、生物学との距離は縮められなかった。酵素の生化学的性質を構造から説明することに力点が置かれ、特異性に関しては大きな成功を収めた。ただ反応活性と機構に関しては、リゾチームとキモトリプシンについての長い歴史に象徴されるように、成功は限定的であった。この時期に、技術的にはX線結晶構造学、NMR構造学とも長足の進歩を遂げ、原子分解能での構造学の標準技術としての地位を確立した。
生物学としての復権は、調節ネットワークの要のひとつであるDNA・蛋白質複合体形成の特異性を、アミノ酸残基と塩基との相互作用として記述することからなされた。このことは、生体高分子の構造をもとにした生物学のパラダイムが変貌しただけでなく、構造生物学が、問題提起よりも手段として生物学に認知されたことを意味する。当時既に生物学全体を裏打ちする技術体系と変貌していた分子生物学と同質となったのである。この変化は、その後のシグナル伝達への貢献においても証明された。日本では研究費配分制度の欠陥のためにこのパラダイムシフトの認知が遅れ、現代の構造生物学の確立において遅れを取ってしまったことは残念な事実である。
90年代前半には、構造研究の成功例から、相互作用の存在と構造モデルは必須であっても、X線やNMRの与える原子分解能自身が生物学に必ずしも必要でない場合があることが明らかになってきた。高分解能のみでは必ずしも生物学に結びつかない場合があるという、構造生物学のこの第2のパラダイムシフトは、電子顕微鏡等の在来技術や、走査プローブ顕微鏡、新しい光学顕微鏡および光学技術による生物学を生み出しつつある。これらの新手法は、X線やNMRの狭義の構造生物学に取って代わるものではなく、それらと補完的な関係にあり、分子の動きと空間配置を測定することにより生理的な調節メカニズムに迫ろうとするもので、1分子計測やナノバイオロジーとして新しい構造生物学を形成しつつある。また、この分野には、生物学に対する問題提起能力を持つものもある。このように、構造生物学には二つのパラダイムシフトがあり、それは構造生物学の定義と目標の変遷だけでなく生物学への寄与の変化でもある。二つ目の新しいパラダイムシフトによって生物学での深い問題提起を行うように変化しつつあることから、構造生物学は脳研究と共に、科学としての生物学医学分野の二大フロントを形成しつつあると言って過言ではない。第一のパラダイムシフトに遅れを取った苦い経験に学ぶ、新たな自覚が必要とされている。
1.2.ゲノム科学進展と構造生物学
ゲノム計画は、一般に誤解されている様に夢の生命科学ではなく、あくまで染色体の物質的記述であり、それ自体が科学として自立している訳ではない。遺伝子の塩基配列から生体の調節や機能へと至るポストゲノム科学が確立しない限り、ゲノム科学としての確立も無く、膨大な情報と巨大な投資が水泡と化す危険に瀕しており、それゆえにポストゲノム科学の重要性と責任を倍加している。そしてこのポストゲノム科学の中核をなすものが構造生物学である。
蛋白質遺伝子の塩基配列から立体構華を求めることは急務であるが、ある程度の目途が付きつつある。構造データの量的蓄積により、いくつかの帰納的方法論が成功をおさめつつあり、この点で構造生物学の寄与は大きい。しかし当初目指された、塩基配列からの一般的な演繹的方法論に関しては、未だ目途は立っていない。
ポストゲノム期の大きな問題の一つは、構造から機能への道である。演繹的手法は大変困難である。例えばドラッグデザインにおける構造からの演繹的手法は、提唱されてから30年以上経つにも関わらず、ランダムに試行を繰り返す帰納的方法の成功に圧倒されている。現在のところ帰納的方法がより確実な方法であるため、多数の蛋白質の立体構造決定をもたらす網羅的研究が必要とされている。この必要性は現実のものであり現在声高に叫ばれているが、立体構造決定自身は機能決定への実行可能な一過程であり、これが解決しても目標が到達されるわけではない。現在のコストパフォーマンスには大きな問題があり、抜本的な改善が急務である。それと並行して、立体構造から機能への帰納的手法に関するオリジナルで多様な研究や、1次構造から機能への立体構造を介しない直接的帰納的方法の開発等が急務である。蛋白質単体から相互作用への、構造生物学のパラダイムシフトで明らかになったように、蛋白質機能自体の研究は、それだけでは生物学ではない。この点にポストゲノム期の最大の問題が潜んでいる。例えば比較的単純なバクテリアの場合でも、塩基配列からそれによって決定されているはずのプロモーターを予想することすら、実用にはほど遠いのが現状である。蛋白質構造の網羅的研究のみにこだわりすぎると、ポストゲノム研究が蛋白質構造研究に歪む可能性がある。
従って、ポストゲノム期の構造生物学に現在緊急に要求されることは、ゲノム情報を取り込んだ分子生物学・細胞生物学等の多様な分野で、独創的な着想に基づく研究が広範囲かつ量的になされることである。この中で初めて、立体構造から機能への演繹的手法の可能性と帰納的手法の実用化が正面から目標とされるとともに、ゲノム情報を調節や細胞機能として生かせる道が開けるのである。
1.3.構造生物学がもたらすもの
現代の生物学は、分子生物学が確立した「表現型の遺伝子による記述」を基本パラダイムとして展開している。このことは本来、遺伝情報(塩基配列)→生体高分子の化学構造→生体高分子の立体構造→分子間相互作用→…→表現型(高次機能)、という階層性をもつはずだが、分子生物学は、最初と最後以外をブラックボックスとして、「遺伝情報→高次機能」と捉え成功を収めてきた。分子生物学はこのパラダイムのもと、いろいろな高次機能に参画している生体高分子を同定し、それらの分子を役者とするストーリーを百科辞書的に記載することに成功してきた。
しかし、複雑で多様な百科辞典的知識の集積は、それらの一般化、普遍化、より深いレベルからの統一的理解への欲求を生みだした。X線結晶構造解析法とNMRディスタンスジオメトリー法の成熟は、ふたたび生物学者の目を、生体高分子の構造へと向けさせた。そして、蛋白質による核酸の特異的認識、シグナル伝達のカスケードとネットワークの構造的理解、免疫機構の物質的理解等に果たした、構造生物学の寄与はあまりに巨大であり、生体高分子を遺伝子の名前だけにとどめることで、生命現象が理解できると考える生物学者は、もう存在しない。
従って、現代の構造生物学は途中を飛ばして成功した古典的な分子生物学のパラダイムに、「生体高分子の立体構造」を挿入することにより、新しい生物学像を構築しようとするものである。構造生物学の任務の一つとして、生命現象を司る分子機械の作用機構を、その立体構造を通して明らかにしようという必然的なものがある。この方向の構造生物学の価値は、80年代以後、不動の価値を持ち続けている。
このレベルでの生物機能研究は、物理学・化学に基づいて生物機能を理解しようとする立場でもある。生体高分子もあくまで分子であるので、全ての分子機械は、ミクロな環境において強い熱雑音に晒されつつ確率的に働くものである。この本質的な性質と、分子生物学が印象づけた決定論的色彩とのギャップがどう埋まっていくのか、という一般的命題が解かれる日も近い。
最近、大きく生物学が動き始めた。時間や空間の概念と遠かった分子生物学では、発生学や脳神経科学のもつ時間的調節、形態的・空間的調節が扱えないと言う困難が、細胞生物学レベルから自覚され始めたのである。ここで新たな構造生物学が必要とされ始めている。第一の理由は、構造こそが形態的・空間的変化を理解できる鍵であることはもちろんである。もっと重要な理由は、時間的調節、形態的・空間的調節を理解するためには、物理学・化学に基づいて生物機能を理解しようという立場に立たざるを得ないことである。一方、要求されている構造生物学は、蛋白質の原子分解能をもつ構造ではなく、むしろ、分解能はもっと低くても妥協出来るが、動的、空間的な性質は妥協できないものである。この方向から生み出されてきたものが、1分子計測や電子顕微鏡、走査プローブ顕微鏡、光学的新手法による新しい構造生物学である。
幸いなことに、この分野で日本の研究は世界をリードしており、新しい構造生物学の名前として、「ナノバイオロジー」という命名をしたのも日本の研究者群である。この分野は、まだ開拓段階であるが、確実に生物学の新しいフロンティアを形成しつつある。
このように構造生物学は分子生物学のパラダイムを乗り越える新しい生物学をもたらすことが原理的には期待される。予測できる構造生物学の近未来的成果の一つは、ゲノム情報の利用法の構造への確立であろう。この成果は、40年来の困難な蛋白質構造論の発展に依存している。現在爆発的に進展しているゲノム科学の結果、分子レベルの生物学研究対象の爆発的拡大は不可避的に起こるであろう。また、その結果得られる多様なデータを比較・分類することによって得られる経験的知見が、基本的な概念構成上も、応用上も、大きな意義をもってくるものと思われる。応用上の意義は、大きな研究費の流入を促し、その結果研究対象の拡大は一層促されるであろう。このポジティブフィードバックは、基礎研究にとっても望ましい。構造生物学の望ましい展開のためには、基礎と応用との望ましい相互作用とバランスに十分配慮する必要がある。
しかし、この側面は構造生物学がもつポテンシャルの一つに過ぎない。本来、構造生物学はゲノム計画に依存して成立しているわけではなく、生物界でおこる現象の理解、つまり様々なレベルの構造を用いてその現象のメカニズムを記述することにある。ゲノム計画の構造生物学への依存は強まるであろうが、構造生物学は本来独自の発展が見込まれる分野である。今後の細胞生物学的方法の発展は、まさに構造生物学の発展が担っている、といえよう。
![]()
2.立体構造を解明する実験的手段
2.1.X線結晶構造解析
X線結晶構造解析は結晶にX線を当てて得られる回折像に基づいて、結晶中の電子密度分布を決定する。この方法では、蛋白質結晶中の原子座標を10分の1Åの精度で決定することができ、対象とする分子の分子量は500万を越えても可能であり、実質的に分子量限界がない。最も多い分子量が10万以下の蛋白質では、その構造解析法もルーチン化されており、他の構造解析法に比べて初心者も容易に利用できる手法である。
X線結晶構造解析では、回折斑点の強度から結晶構造因子の絶対値を求め、別に決められる位相と組み合わせてフーリエ変換によって結晶中の電子密度分布を求める。蛋白質結晶構造解析の最も克服すべき困難な課題は、如何にして位相を決定するかという位相問題であった。重原子同型置換法がこの問題を克服する主力になっている。この方法では、構造決定すべき結晶の他に、この結晶に重原子を導入した結晶を作り、両者の回折強度分布の差に基づいて位相を決定する。分子量が大きくなると重原子を導入しても回折強度の変化は大きくならず、この方法による位相決定が困難になる。ところが、強力で平行性の高い放射光X線の出現によって、精度の高い回折強度測定が可能になり微小な強度差も検出できるようになった。その結果、現在500万を越える分子量の位相決定もこの方法で可能になっている。
放射光は異なった波長のX線を自由に取り出せるために異常散乱を利用した位相決定法を可能にした。この方法では、標準波長のX線の他に特定の原子によるX線の散乱能が変化した波長のX線による回折強度を測定する。同型置換法と同様にこれらの回折強度の差に基づいて位相を決定する。金属などの重原子を含む蛋白質ではその結晶だけで構造決定が出来る。メチオニンの代わりにセレノメチオニンを導入した蛋白質を容易に作り、構造決定することが、ルーチン的に行われている。さらに、Xeなどの希ガスを結晶にドープすることにより、重原子置換体を得る方法が開発され、一定の成果を収めている。
もう一つのよく使われている位相決定法には、既に決められている類似の構造を元にし 、て位相の近似値を求め、順次精密化する分子置換法がある。この方法は簡便で、微小な構造の変化と機能や物性の関係を系統的に調べる研究によく使われている。
単結晶を用いた構造解析では静的構造しか求められないと思われていたが、放射光によって連続X線も使えるようになり、古くからあったラウエ法が蛋白質の構造解析法として現実のものになった。単波長のX線による回折実験では結晶をあらゆる方位に設定して回折像を測定する。それに対してラウエ法では連続した広い波長範囲のX線を用いて1ないし数枚の回折像の測定を行う。従って短時間で回折実験を完了することが出来るので、立体構造の時間変化を追うことが出来る。この方法による動的構造解析は、蛋白質X線結晶構造解析に新しい可能性を与えている。
X線結晶構造解析は、生体分子の立体構造決定法として不動の地位を確立しているが、ここに至るまで位相問題以外にも様々な問題が提起された。一つは、結晶状態の構造を決めるために、「結晶中の構造は溶液とは同じである筈がない。従って結晶構造解析による構造で生体内の現象を議論することは出来ない。」という指摘であった。これに対しては、結晶化した蛋白質が酵素活性を示すことによって反論した。また、結晶中では平均で半分の空間は溶液状態と同じ溶媒によって占められており、有機化合物の結晶のように分子の立体構造に影響を与える分子間の相互作用は極めて少ないことも明らかになった。NMRによる溶液状態の構造ともよく一致することからも、結晶状態と溶液状態の立体構造の同一性は実証されている。
結晶を用いることがこの方法の弱点と見られることがしばしばあるが、この方法が精度の高い構造決定法であることを考慮すれば、結晶を利用することは最善の手法である。なぜならば、精度を上げるためには“実験データ”の正確な平均操作を行うことが不可欠である。結晶はハード的に正確な平均操作を自動的に行っている。この意味でも結晶による回折法は正確な構造決定法として普遍性を持っている。
この手法に対する注文は、数mgを越える大量の試料が必要なことである。この問題を結晶学が解決出来ないでいる間に、DNA技術の進歩が蛋白質の大量調製を可能にした。蛋白質の機能の研究は結晶化とはけた外れに少ない量で行えるようになっているが、蛋白質の物性などを調べるためには結晶化と同等の量が必要である。蛋白質の大量調製は結晶構造解析のためのみならず、蛋白質の本質を理解するために避けて通れない課題である。
現在、蛋白質結晶学はより正確な構造解析、より複雑で大きな分子の構造解析、より多様な構造を求める構造解析の3つの座標軸を持つ空間で進歩を続けている。いずれに対しても放射光とDNA技術は飛躍的進歩をもたらしたが、まだまだ手が及んでいない領域も多い。例えば、膜蛋白質は生体内の蛋白質の30%にも達するのに、構造解析されたものは30種に満たない。大量調製とそれに続く結晶化が依然として蛋白質結晶学の抱える最大の課題である。
2.2.NMR
NMRは溶液状態の試料を測定対象とする溶液NMRと、粉末あるいは結晶・液晶状態の試料を用いる固体NMRに大別される。現時点で構造生物学への貢献が大きい溶液NMRから得られる構造情報をまとめると以下の様になる。
2.2.1.生体分子の原子レベルでの立体構造決定
蛋白質や核酸といった生体高分子の立体構造決定を行うことが出来る。従来は主として核オーバーハウザー効果(NOE)から得られる原子間の近距離情報のみを構造情報として使っていたため、構造決定の精度はあまり高くなかった。ところが最近、原子間の双極子−双極子相互作用、化学シフト、水素結合を介したスカラー結合、異種核間のスカラー結合、回転拡散の異方性などから得られる情報が立体構造解析に用いられるようになり、一般的なX線結晶法に匹敵する精度での構造解析が可能になってきた。
NMR法による立体構造解析の利点としては、2万ダルトン程度の比較的小さな分子の解析が迅速である点が挙げられる。また対象分子の溶液でのスペクトル測定は簡単に出来るので、それが構造解析可能であるかどうかのチェックは容易である。こういった点は蛋白質ドメインなどの網羅的な構造解析に向いていると思われる。
一方、欠点としては、解析分子量に上限がある、安定同位体ラベルが必要である、が挙がられる。解析分子量の上限は45,000程度であるが、実際の解析は25,000を超えると困難になる。最近のTROSY法などの開発に伴ってこの上限は伸びつつあるが、X線結晶法で可能な10万−100万ダルトンを超える超分子系の立体構造解析を可能にする手法の開発のめどはたっていない。
2.2.2.生体分子間の相互作用の解析
生体分子間の相互作用部位の迅速な解析はNMRの得意とするところである。特に15N核で安定同位体ラベルされた蛋白質試料を使った、蛋白質−蛋白質、蛋白質−核酸、蛋白質−低分子間相互作用の解析は広く行われている。解析手法としては、化学シフトパータベーション法、H-D交換法や最近開発されたスピン拡散法などが挙げられる。
また最近開発されたスカラー結合測定により水素結合を同定する手法は分子間水素結合の解析を可能にする。こういった手法は分子間相互作用の網羅的な解析を可能にするため、いわゆるポストゲノム(シークエンス)期の蛋白質機能解析では、重要な役割を果たすと期待される。
2.2.3.蛋白質の運動性の解析
蛋白質の回転・併進といった外部運動や、ピコ秒からナノ秒オーダーでの内部運動の定量的解析が可能である。特に内部運動に関しては、分子全体に及ぶ残基レベルの情報を与える唯一の方法であり、NMRのこの分野に与える寄与は極めて大きい。またマイクロ秒よりも遅い運動に対しても半定量的な知見を与え、幅広い時間領域での情報を与える手法となっている。このような手法は最近、分子間相互作用において運動性が減少する場合のみならず、増加する場合があるという興味深い現象を明らかにした。蛋白質の運動性は今後、その機能発現を理解するのにますます重要になると思われる。また蛋白質ほどではないが、核酸の運動性の研究も行われている。
2.2.4.蛋白質のフォールディング
フォールディング中間体について分子全体で残基レベルの情報を与えるため、重要な手法となっている。特に化学シフト値のみから2次構造が推定できるため、フォールディング時の2次構造形成順序や時期について、他の手法では得られない重要な知見を与える。
2.3.電子顕微鏡
2.3.1.電子顕微鏡による構造研究の特徴
膜蛋白質などの複雑な生体高分子やその複合体の構造研究は生物学的な重要性にも関わらず容易ではない。ゲノムプロジェクトの進展で、原核生物から高等動物にいたるすべての生物において膜蛋白質は全蛋白質の30%をしめることが明らかになってきているが、現在までに発表されている構造解析された膜蛋白質の種類は20程度と極めて少ない。このような膜蛋白質の構造研究には電子線結晶学が有力であることが証明されつつある。電子線結晶学を用いると、2次元結晶の質によるが、生体膜の中にある状態に近い条件で、3Åを越える分解能で膜蛋白質の構造を解析できる。これ以上の分解能の解析では脂質分子も明瞭に見ることが出来る。
さらに、電子線に対する原子散乱因子は、原子が電荷を持たない場合とイオン化している場合では大きく異なる値を示す。それゆえ、電子線結晶学ではX線と異なり蛋白質の構造における電荷の移動や、イオン化の状態を直接研究できる可能性がある。具体的にはポリペプチドのカルボニル基において、およそ40%程度の負の電荷が酸素に移動して、炭素が同程度正に帯電しているというようなことが実験的に観察される。また、結晶を作製できないような複雑な蛋白質、微量にしか精製できない蛋白質やその複合体などについても電子顕微鏡を用いた単粒子解析法によって、立体構造が解析できる。多くの粒子の画像を解析することによって2次構造を議論できる分解能、すなわち、7Aの分解能での解析ができるようになってきた。この手法を用いて、原理的には原子レベルの分解能の解析が出来るとされているが、現在のところそのレベルの解析はなされていない。チューブ状の結晶やらせん対象を有する試料については対称性のない試料より高い分解能の解析が可能である。特にチューブ状結晶を用いると原子レベルの分解能での解析が可能になってきた。
トモグラフィーという手法を用いることによって、さらに複雑な蛋白質複合体や細胞組織等の立体構造を研究できる。ただし分解能は数10A程度であり、原子モデルが出来るような解析は不可能である。ただし、生物学的に興味深い生体高分子複合体の構造研究の1つの手段として、特に米国などでは注目されてきている。
2.3.2.世界および我が国の状況
膜蛋白質の構造研究は、世界の研究レベルと対等に進んでいる。重要な膜蛋白質が日本人が関与した研究で解析されてきたし、されつつある。中でも電子線結晶学を用いて解析された膜蛋白質は3つから4つ程度になろうとしているが、これらすべてに日本人が関与している。電子線結晶学は英国ケンブリッジ、MRC分子生物学研究所で開発され発展してきた手法であるが、日本は試料作製法、極低温電子顕微鏡およびその周辺技術、解析プログラム等すべての面で世界的なレベルにある。ただし、2次元結晶を作製することが高分解能の構造解析には必要であるが、これは3次元結晶作製技術の円熟度から見ると大きく遅れている。世界的にも結晶化できる量の膜蛋白質の精製と2次元結晶作製がボトルネックとなっている。
単粒子解析は欧米で進んできたもので、残念ながら日本は遅れている。ただし、電子顕微鏡や試料作製法では優位に立っているので、今後の展開次第では世界に追いつける可能性はある。ところが電子線結晶学も含めて研究組織が少なく、研究者も少ないので、最近非常に多くの研究グループを増設している欧米に追いつくことが出来るか不安である。トモグラフィーなどは欧米の進歩に比べると全く遅れていると言わざるを得ない。ただし、らせん対象を有する試料の解析については世界的なレベルにある。
2.4.中性子散乱・回折
2.4.1.中性子散乱・回折の特徴
中性子の散乱回折現象を利用して、生体高分子の構造を解析することができる。その原理は、X線結晶構造解析やX線散乱と変わるところはない。X線が電子により散乱されるのに対し、中性子は核力により原子核によって散乱される。したがって、X線の原子散乱因子に見られるような顕著な原子番号依存性はなく、Hのような軽原子をも見ることができるという利点がある。さらに、X線では見られない同位体効果という特徴もある。これらの特徴により、構造生物学における中性子利用の魅力は2点ある。一つは、HとDの散乱長の大きな違いを利用するところであり、もう一つは準弾性散乱・非弾性散乱の利用である。
結晶構造解析において、中性子の有利な点は水素が見えると言うことである。機能との関係で考えれば、解離性アミノ酸の解離基の水素や水和している水などが特に重要である。X線結晶構造解析では見ることのできないこれらの水素が見えれば、機能発現の分子機構の理解が一層深まる。さらに、様々な基質類似体を用いた中間状態の構造解析や時分割中性子結晶構造解析が可能になれば、機能発現中の水素の動きが観測できることになり、その影響の重大さははかりしれない。
HとDの散乱長の違いは、結晶構造解析以外にも、溶液散乱でも有効に応用される。溶媒のH2O/D2O比を変えることで、構造情報量を3倍に増やすことができる(コントラスト変調法)。また、二成分系においては一方の成分の平均散乱長に溶媒の散乱長を一致させればその成分の寄与を消すことができる。例えば、遺伝情報の伝達過程は、核酸と蛋白質の織りなす極めて協同的で複雑な反応過程である。この様な系でコントラストを変えて中性子散乱を行えば、蛋白質からのシグナル、核酸からのシグナル及び相互作用に関わるシグナルをそれぞれ分離して得ることができる。中性子以外の方法で、このような解析は行えない。中性子は、複雑な系での(しかも生命現象の理解にとって最も本質的な系での)測定に威力を発揮する可能性を秘めている。特定のアミノ酸をD化した変異蛋白質を得ることができれば、そのアミノ酸の分子内での分布を調べることもできる。
X線で行うことのできない実験が、準弾性・非弾性散乱実験である。中性子準弾性・非弾性散乱は蛋白質の動力学についての情報を与える。その実験結果は、直ちに分子動力学計算や規準振動解析などの理論計算の結果と比較することができる。特に低エネルギー領域の動力学の重要性が指摘されているが、中性子非弾性散乱は、蛋白質の機能と結びついた低エネルギーの動的性質を理解するための、極めて優れた測定手段である。また、機能を発現できる状態の蛋白質の動的性質は線形調和的ではないことが知られ始めた。蛋白質の動力学の非調和的な側面の測定は調和的な(振動的な)測定に比べると非常に困難である。蛋白質の非調和的な運動は中性子準弾性散乱として観測される。しかも、Hの寄与が圧倒的であるため、全ての蛋白質について適用可能である。従って、蛋白質の動的性質を調べるために、中性子は不可欠の手段となる可能性を秘めている。
中性子散乱・回折に用いる中性子のエネルギーは80meV以下と小さいため、中性子による試料の放射線損傷は殆ど無く、結晶構造解析や非弾性散乱のような長時間実験にも十分耐えられることは試料の取り扱いにとり大変有利である。
2.4.2.近年のわが国と世界の状況
わが国で世界に先駆けて開発実用化された中性子イメージングプレート(NIP)は従来の中性子蛋白質結晶構造解析を根本から変えた。従来の方式では構造解析用データ収集に通常の原子炉を用いた実験で約100日のマシンタイムを要し、しかも得られる構造精度も悪かった。これは現実には不可能を意味しており、事実、ここ15年近く実験は行われてこなかった。NIPの実用化により、データ収集速度が10倍に且つデータの精度も格段に向上した。原研ではNIPを用いた中性子回折計(BIX−3)を建設し、すでに2,3の蛋白質分子の解析を1.5オングストローム分解能で行っている。BIX−3は世界トップの性能を有する装置として認められている。最近、リゾチーム蛋白質の全ての水素と、水和水が観測され、特に水素結合については、これまでの定説に疑問を投げかけるような結果が得られている。ただし、中性子の場合、1−2mmの大きさの単結晶が必要である。しかし、決して不可能なことではなく、多くの蛋白質の中性子解析による研究が始まっている。
溶液散乱については、ある程度汎用の測定法としての地位を占めてきている。しかし、ビームタイムの慢性的不足から、興味深い結果がなかなか生産されていない。非弾性散乱については、わが国には、満足なデータをえることのできる装置が皆無である。測定のためには、フランスのラウエランジュバン研究所やイギリスのラザフォードアップルトン研究所にでかけなければならない。ラザフォードアップルトン研究所には世界最強のスポレーション中性子源があり、反射率計による生体膜中の分子の構造研究等で成果が上がっている。しかし、蛋白質の動力学の理論的研究では日本は世界のトップクラスに位置している。蛋白質の中性子準弾性・非弾性散乱の実験研究は始まったばかりであり、優れた測定がなされれば、トップレベルにある理論家との共同作業により日本はこの分野で世界をリードする可能性が強い。
2.4.3.中性子源将来計画
研究用原子炉を用いての中性子源はグルノーブルの高中性子束原子炉を最後に技術的問題を克服出来ず実現していない。一方、1970年代後半から陽子加速器を用いた核破砕中性子源が建設され始め、遂に約10年程前にグルノーブルの高中性子束原子炉に匹敵する核破砕中性子源がRALに完成し、将来の中性子源計画はすべて核破砕中性子源に取って替わった。米国では昨年度より2MW核破砕中性子源(RALの10倍)の建設が開始され、2005年完成予定である。ヨーロッパは机上プランの段階である。
日本では高エネルギー加速器研究機構と日本原子力研究所の共同建設として、1MW(引き続き5MW増強)の核破砕中性子源計画があり、すでに予備調査費が計上されている。構造生物学は、ここの中性子散乱を用いる研究の主要な柱と位置付けられている。現在のグルノーブルの高中性子束原子炉と比較すると、単結晶構造解析では約0−100倍、小角散乱では5−10倍、非弾性散乱では500−1000倍の強度が予想されており、中性子構造生物学の様相が大きく変わると期待される。
2.5.X線繊維回折・溶液散乱
X線の回折・散乱現象を利用して生体高分子の構造情報を得る最も洗練された方法は結晶構造解析法であるが、この方法を適用するためには、対象物質の単結晶が必要となる。一般に生体内で、タンパク質が3次元結晶を構築することはほとんどない。多くのタンパク質は水溶性であり、細胞内や外の水溶液環境に存在している。溶液中でのタンパク質の構造情報を得る手段がX線溶液散乱である。一方、生体中において低次元の規則構造は比較的よく見られる。例えば、筋肉や髪の毛などはタンパク質が繊維状に集まった集合体であり、繊維軸方向には規則性がある。細胞を取り囲む細胞膜や細胞内小器官の膜は、二次元の集合体である。繊維状物質の構造情報を得る手段がX線繊維回折である。また、膜についても回折によって膜厚方向及び膜面内に関する構造情報を分離して得ることができる。
対象物質が繊維状でありらせん対称を有している場合、高配向度の試料により、原子レベルでの分解能の構造解析が可能である。この方法で、タバコモザイクウィルス、バクテリアの鞭毛などの構造解析がなされてきた。また、原子レベルには達していないが、筋収縮の分子機構を明らかにするための筋繊維の構造解析も成功を収めてきている。特に、筋繊維については、時間分解測定も積極的に試みられている。放射光を利用することにより、繊維回折は今後ますますその威力を発揮していくと期待される。アミロイドなど病因となる組織内の繊維状物質の構造解析も、この手法によって可能になるであろう。
溶液散乱は、分子のサイズや形状が与えられるだけであり、分解能の低い構造研究法である。しかし、蛋白質溶液があれば測定できるという簡便さと、どのような溶媒条件下で測定が可能であるという汎用性から、広い範囲で利用されるようになってきた。特に、折畳み中間体などの非天然状態の構造研究への応用が、この手法の有用性を高めた。また、結晶構造データから溶液散乱プロファイルの計算が簡単にできるため、結晶構造と溶液構造の比較や、リガンド結合などに伴う構造変化の推定などに積極的に利用されている。また、ストップトフロー、温度ジャンプあるいは閃光照射などを組み合わせることで、時間分解測定が可能となり、機能中の蛋白質の構造変化を追跡することができる。実験結果の解析においては、分子動力学シミュレーションと組み合わせることで、その有効性は増す。
我が国においては、繊維回折、溶液散乱ともに、高いレベルでの研究が行われている。
2.6.走査プローブ顕微鏡、新世代光学顕微鏡
走査プローブ顕微鏡は、1981年に発明された走査トンネル電流顕微鏡(STM)に端を発し、様々な方式のものが開発された。原子分解能を持つもので、もっとも普及しているものは原子間力顕微鏡(AFM)である。蛋白質の構造解析に試用されたこともあったが、現在の用途の多くは、電子顕微鏡よりも簡易なイメージの取得法として、蛋白質によるDNAの曲がりの検証など、0.1−1000nmの分解能をもって用いられている。プローブの直径分(10−100nm)像が太るのが欠点であるが、逆に高さは、実寸が測定できる。水溶液中でも用いることが出来るので、生体試料に適していると思われるが、表面に固定された分子を対象とするため、水中AFMはイメージとしてよりも、ゆっくり動く2次元平面内のDNA蛋白質相互作用の動的解析などにその力を発揮している。
表面固定と熱雑音を除いた、クライオAFMは、実用化直前であり、非染色・非固定で原子分解能を出す手段として期待されている。また、フリーズドフラクチャーとAFMの組み合わせは、電顕の様に熟練した技術を要求しない高分解能技術となっている。この他、光学プローブを持つ近接場顕微鏡やマックスウェル応力顕微鏡なども実用化段階に入りつつあり、守備範囲の広いイメージング技術として、1分子DNA塩基配列決定などに挑戦が続けられている。
また、AFMは、化学で記述されていた現象を、力学で記述し直すことにより、より機械論的立場に立った相互作用の理解を可能にした。この点で、原子分解能で熱揺らぎより小さな力を測定できる光フィードバックによる超高感度AFMは、日本のグループの発明であり、将来が期待されている。
ビデオ増強顕微鏡(1981)、共焦点顕微鏡の登場(1983)以来、光学顕微鏡技術の進歩は、構造生物学の技術的進歩の中で、より分子生物学・細胞生物学に密着したものである。レーザートラップ、2光子励起や日本の発明である光センシングナノメトリーなどと、標準技術になりつつあるGreen
Fluorescent Protein(GFP)によるin vivo蛍光ラベルなどとの組み合わせにより、光学顕微鏡の広い応用が可能になり、特に1分子計測には、新しい相互作用測定技術の一部として、組み込まれている。電子顕微鏡と同様に光学顕微鏡も、かつては形態学的手段であったものが、新技術により構造生物学的手段に脱皮しつつある。顕微鏡本体の改良も白色共焦点顕微鏡や生体内の物質の濃度を測定できる新たな蛍光顕微鏡の開発など野心的な取り組みがなされている活気のある分野である。ヨーロッパには、これらの新技術の集積場所として、光学顕微鏡センターが近年設置された。
![]()
3.立体構造データベース
3.1.世界およびわが国における現状
世界中の構造生物学の研究者が決定した様々の蛋白質の立体構造は、蛋白質立体構造データベースに整理されて蓄積されてきた。
歴史的には、PDB(Protein Data Bank)と呼ばれるデータベースが、1971年に誕生して以来、米国Brookhaven
National Laboratory(BNL)が管理・運営を1999年5月まで継続してきた。しかし、1999年6月から、Rutgers大学、San
Diego Supercomputer Center(SDSC)、National
Institute of Standards and Technology(NIST)の3者が協力して運営するResearch
Collaboration for Structural Bioinformatics(RCSB)という組織が、BNLに替わって管理・運営を開始した。(http://www.rcsb.org/pdb/)
このデータベース維持のため、RCSBはNational
Science Foundation(NSF)から1千万ドル、5年間のグラントをもらい、Rutgersに13名、SDSCに11名、NISTに8名、総勢32名の規模で、プロジェクトを進めている。RCSBにおけるデータベース運営の最も大きな特徴は、Rutgers大学、SDSC、NISTという3つの部署に作業を分担したことにある。具体的には、Rutgers大学においてデータの受理と提出されたデータの編集を行い、SDSCにおいてデータの統合化と配付を行い、NISTはデータベース全体の監督と公文書化の作業を行っている。このデータベース運営の分業化は、データベース運営に関して多くの利点があり、見習うべき所が多いと思われる。すなわち、立体構造データベースにおいて欠かせない、理学の専門家によるデータのreviewingと、情報科学の専門家によるコンピュータとネットワークによるデータベース管理・配付とを、それぞれの専門家集団がいる場所に分けてしまっている点である。RCSBでは、このように管理・運営を合理化したのみでなく、データ・フォーマットの変更、自動登録ソフトの開発など、登録者および閲覧者の利用の簡便化等を進めている。
図に示すように、データの量は近年急速に増加し、2000年4月には1万2千件近い量の立体構造データが登録され、公開されている。登録数は、米国からは約50%、ヨーロッパからは30%、アジアからは10%でその半分ほどが日本からだと言われている。具体的には、1999年のAutoDepによる自動登録の全体数2090件のうち、アジア・オセアニア地区からは287件(14%)、その内日本からは133件(6.4%)であった。また、2000年の第一四半期では、同様に自動登録全数587件のうち、アジア・オセアニア地区からは89件(15%)、その内日本からは43件(7.3%)であった。
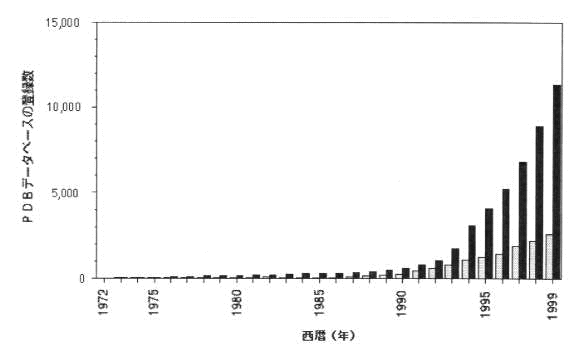
図3.1.蛋白質立体構造データベース(PDB)に登録されているデータ数の変遷。白棒が、各年毎に登録されたデータ数であり、黒棒は積算総数を表す。
一方、EBI(European Bioinformatics Institute)では、MSD(Macromolecular
Structure Database)プロジェクトというのを、PDBとは独立したデータベース管理システムとしてスタートさせている(http://msd.ebi.ac.uk/)。1996年9月から英国HinxtonにあるEBIのDr.Goeff.
Bartonがヘッドとなった5名のスタッフにより、従来のPDBデータを独自の新しいフォーマットに変換・整理し、さらに新しい立体構造データの登録受付を行っている。既に1998年からは、毎月50件ほどの登録を受け付けているという。PDBとは協力してデータの共有をはかっていて、どちらかに登録すれば双方のデータベースに同時に登録される。このプロジェクトは、将来的には12名ほどのスタッフにより、年間100万ポンドほどの予算で運営することを目指している。
日本では、大阪大学蛋白質研究所が、米国PDBデータベースのアジア・オセアニア地区での公式のアーカイブとして、米国ラトガース大学/米国カルフォルニア大学サン・ディエゴ校のグループと協力してデータベース管理・運営を行っている。インターネットを通して、このPDBデータベースにアクセスできる(http://pdb.protein.osaka-u.ac.jp/pdb/)。また、上記したデータ量の急増に対処するため、国内はもとよりアジア・オセアニア地区の構造生物学者が解析した立体構造情報の新規登録作業を、米国ラトガース大学と協力して開始しようとしている。
ところで、NMRによって決定された立体構造もPDBには登録されているが、単なる分子の構造以外にも、各蛋白質中で同定された炭素、窒素、水素の各スピンの化学シフト情報が、Bio
Mag Res Bank(BMRB)というデータベース(http://www.bmrb.wisc.edu/)に集積されている。これは、Wisconsin大学のJohn
L.Markleyによって1996年からスタートしたものであるが、PDBおよびEBIとも協力し、平成12年4月末においては、総エントリー数1568件のうち、蛋白質・ペプチドの1547件に対して1H化学シフトは203,672件、13Cは59,707件、15Nは22,991件である。他は、DNAおよびRNAの化学シフト値である。このデータベースに対しては、日本国内で対応し国際協力をしている所が未だになく、早期に対応すべきだと考えられる。
3.2.バイオインフォーマティクス現状と動向
ゲノム・遺伝子の情報は莫大な量となりつつあり、コンピュータの助けなしでは、これらの情報を解釈し生命現象を理解していくことは困難となっている。この目的のために、数年程前に情報科学と生物学が結びつき、ゲノムの塩基配列情報や蛋白質立体構造情報をコンピュータ処理し利用する科学・技術としてバイオインフォーマティクス(「生命情報科学」と訳される)という学問が誕生し発展した。現在、世界各地で、ゲノム配列や蛋白質立体構造データベースを維持・管理する所を中心として、多くのバイオインフォーマティクス研究がなされ、蓄積されたデータからのデータマイニング、およびそのためのツールとしてのソフトウェアが開発されている。また、特に米国では、ベンチャー企業が、特化したデータベースや技術を開発し、活発なバイオインフォーマティクス研究が進められている。
バイオインフォーマティクス研究には、配列比較から立体構造を経由せずに、各遺伝子に対応する遺伝子産物の機能情報を得ようとするFunctional
Genomics(機能ゲノム科学)研究の一環としてのものと、立体構造情報から各遺伝子産物の生化学機能を論じて生物学的機能へ至ろうとするStructural
Genomics(構造ゲノム科学)研究として位置づけられるものとがある。後者は、特にStructural
Bioinformatics(構造バイオインフォーマティクス)と呼ばれ、構造生物学の一つの分野を形成しており、次節で詳述する。
生物学関連のデータベースを維持管理し、その活用としてのバイオインフォーマティクス研究を行っている大きな研究グループとして、以下のものが挙げられる(順不同)。
[海外]
・ National Center for Biotechnology Information(NCBI),
USA(MEDLINE, Entrez等の文献データ、DNA配列データ)
・ EMBL Outstation(European Bioinformatics
Institute:EBI), UK(DNA配列データ、立体構造データ、構造比較)
・ The Genome Sequence Data Base(GSDB),
National Center for Genome Resources, USA(ゲノムデータ)
・ University of Washington and the CaPCURE
Genetics Consortium, USA (Expressed Sequence
Tags(ESTs) from prostate cDNA libraries)
・ U.C.Berkeley, Berkeley Drosophila Genome
Project, USA(ショウジョウバエ・ゲノムプロジェクト)
・ Genome Sequencing Center, Washington
University, St. Louis, USA(線虫ゲノム)
・ The Sanger Center, Hinxton Hall, UK(ゲノム)
・ The Genome Dababase(GDB), The Bioinformatics
Centre at the Hospital for Sick Children(HSC)
in Toronto, Canada, and The Johns Hopkins
University Schoo1 of Medicine in Baltimore(JHU)(ヒトゲノム)
・ The PROTEIN INFORMATION RESOURCE(PIR),
a division of the National Biomedical Research
Foundation, USA(蛋白質アミノ酸配列)
・ Munich Information Centre for Protein
Sequences, a bioinformatics group of the
GSF(National Research Center for Environment
and Health) at the Max-Planck-Institut f.
Biochemie, Germany(蛋白質アミノ酸配列)
・ Swiss Institute of Bioinformatics(SIB),
Swiss(蛋白質アミノ酸配列,アミノ酸モチーフデータ)
・ Northwestern University, USA(The Kabat
Database of Sequences of Proteins of Immunological
Interest)
・ MRC Laboratory of Molecular Biology and
Centre for Protein Engineering, Cambridge,
UK(蛋白質構造分類)
・ Biomolecu1ar Structure and Modelling
Unit, University Co11ege London, UK(蛋白質構造分類)
・ Research Collaboratory for Structural
Bioinformatics(RCSB), Rutgers, The State
University of New Jersey, USA(蛋白質立体構造データ)
・ Bio Mag Res Bank(BMRB), University of
Wisconsin-Madison, USA(蛋白質、ペプチド、核酸のNMRデータ)
[国内]
・ 京都大学化学研究所(ゲノム,KEGG:分子間相互作用データベース)
・ かずさDNA研究所(ランソウゲノム)
・ ヘリックス研究所(完全長cDNAデータベース)
・ 国立遺伝学研究所(ゲノム、立体構造予測)
・ 東京都立大学大学院理学研究科生物学教室・進化遺伝学研究室・細胞遺伝学研究室(ショウジョウバエ)
・ 名古屋大学名古屋大学大学院理学研究科生命理学専攻(メダカ)
・ 岡山大学資源生物学研究所(オオムギ)
・ 農水省 農林水産ジーンバンク植物遺伝資源部門(イネ)
・ 九州大学農学部生物遺伝資源情報総合センター(イネ)
・ 九州大学理学部生物学教室細胞遺伝学講座(アサガオ)
・ 宮城教育大学生物学科 仙台シロイヌナズナ種子保存センター(シロイヌナズナ)
・ 奈良先端科学技術大学(大腸菌等微生物)
・ 基礎生物学研究所(大腸菌)
・ 通産省製品評価技術センター(超好熱古細菌)
・ 東京大学医科学研究所ヒトゲノム解析センター(ゲノム)
・ 筑波大学遺伝子実験センター動物遺伝子実験室(細胞性粘菌)
・ 理化学研究所ゲノム科学総合研究センター(マウス/ヒトゲノム)
・ 科学技術振興事業団高機能基盤生体データベース(ヒトゲノム)
・ 国立遺伝学研究所 生命情報研究センター(変異タンパク質データベース)
・ 理化学研究所ライフサイエンス筑波研究センター理研ジーンバンクBioinfo
Bank
(蛋白質熱力学データベース、蛋白質核酸相互作用データベース)
・ 九州大学大学院生物資源環境科学研究科遺伝子資源工学専攻(シグナリングパス・データベース)
・ 大阪大学蛋白質研究所 生体分子解析研究センター(蛋白質・核酸など生体高分子の立体構造データベース)
・ 京都大学大学院理学研究科化学専攻(蛋白質の構造・機能分類)
・ 東京農工大学工学部生命工学科(膜蛋白質データベース、膜蛋白質予測)
・ 東京理科大学生命科学研究所(国際蛋白情報データベース(JIPID))
・ 名古屋大学大学院理学研究科生物学専攻(モジュール解析)
3.3.立体構造データベース利用法の展開
3.3.1.比較・分類
蛋白質立体構造が大量に蓄積されるに従って、タンパク質立体構造内のドメインの構造型(内部の二次構造の配置のしかた・トポロジーを意味し、最近ではフォールドという言葉で呼ばれる)の多様性に対する研究が進んでいる。
登録された配列情報と立体構造情報の量とから、タンパク質のフォールドは約1000種ほどであり、最も多めに見積もっても8000種には至らないと言われている。現状での立体構造データベース登録量の増加ペースや、5.2の蛋白質全ファミリー立体構造決定プロジェクトを考えると、近い将来にはほとんどのフォールドがデータベースに登録されると期待される。
このタンパク質立体構造データに蓄積された情報を整理する博物学が1995年頃からまり、インターネットの発展とともに、タンパク質立体構造を考察する強力なツールとなっている。作成者によってそれぞれ、SCOP、CATH、Dali/FSSP、Entrez/VAST等と命名されて、タンパク質の立体構造分類の研究が発表され、インターネット上に公開されている。
それぞれに、分類を行う手法が異なり、フォールドの名前も同一ではない。また、立体構造情報のみでの分類と、配列情報を加味した分類とにも分かれており、未だに統一的なものにはなっていない。しかし、それぞれ階層的分類がなされている。
SCOP(http://scop.mrc-lmb.cam.ac.uk/scop/)は、英国MRCのA.
Murzinらによって1995年頃に最初に作成・公開された構造分類サイトであり、Class-Fold-Superfamily
-Familyの階層に別れている。9912PDB Entries(1
Nov 1999)に対しては、全部で22140のドメイン構造が、520
fold、771 Superfamilyに分類されている。このデータベースの特徴は、二次構造の並び方やトポロジーによるfoldの分類が本質的に人間の視覚によっていることである。
CATH(http://www.biochem.ucl.ac.uk/bsm/cath/)は、英国University
College LondonのJ. M. Thorntonらによって1997年頃に作成、開発され、class(C)-architechture(A)-topology(T)-homologous
superfamily(H)の4つの階層に分類される。全体は自動的に分類・整理されているものの、二次構造としてのarchitechtureに関しては、未だ人間が手を入れて編集している。
Dali/FSSP(http://www2.ebi.ac.uk/dali/fssp/)は、L.
Holm(現英国EBI)とC. Sanderよってドイツ・ハイデルベルグのEMBLで1996年に作成された、距離マトリックスから最適な対応するペアーを選び出す立体構造アラインメントのプログラムであるDaliを用いた、完全に自動化されたデータベース(FSSP:Fold
classification based on Structure-Structure
alignment of Proteins)である。
Entrez/VAST(http://www.ncbi.nlm.nih.gov/Entres/structure.html)は、米国NCBI(National
Center for Biotechnology Information)のS.
H. Bryantらによって作成され、もともと文献データベースとしてのEntrezに、VAST(Vector
Alignment Search Tool)と呼ばれる、内部の二次構造単位の型や相対配置の類似性を高速に調べるアルゴリズムを用いた立体構造比較プログラムに、立体構造データベース(MMDB:Molecular
Modelling Data Base)を加えて、"Structure
Neighbors"が高速に検索されるシステムとなっている。
SCOP、CATH、Dali/FSSPは、それぞれ人間の介入するしかたに違いがあるが、客観的な比較によって、その2/3は全く同一であることが調べられている。また、これらの分類・構造データベースにアクセスすることによって、既存のフォールドとの一致、不一致を探索することが直ちにできるようになっている。
3.3.2.相互作用の解析
Functional Genomics(機能ゲノム科学)の分野では、蛋白質間相互作用を網羅的にHigh
Through Putで直接観測する手法として、酵母のTwo-hybrid-system(Y2H)が提案され、対象とする蛋白質が関連する分子間ネットワークの同定が迅速に行われ始めている。
一方、Structural Genomics(構造ゲノム科学)の分野では、蛋白質間相互作用を蛋白質複合体の実体として観測、解析する。蛋白質複合体のデータベースとしては、英国EBIのグループによって、PQS(Protein
Quaternary Structure, http://pqs.ebi.ac.uk/)が作成されており、もとのPDBデータを基に、ホモおよびヘテロの蛋白質会合体、複合体の立体構造データを得られ、分子間相互作用を立体的に理解できる。
また、理研の皿井らによって、蛋白質と核酸の相互作用に特化した、構造と複合体形成における熱力学のデータベース(Protein-Nucleic
Acid Complex Database, http://www.rtc.riken.go.jp/recognition.html)が構築されている。
特に抗体・抗原の複合体に焦点を当てたデータベースが、270ヶほどの抗体の立体構造に対し、メキシコのC.
AlmagroらによってABG(Directory of 3D structures
of antibodies, http://www.ibt.unam.mx/vir/structure/structures.html)として作成され、公開されている。
こうした蛋白質立体構造のデータをもとに、分子認識機序を構造面から解析するアプローチが従来から様々になされている。さらには、蛋白質分子表面の解析によって、相互作用が行われる表面の特徴をデータベースから解析するアプローチも行われている。
ところで、抗体タンパク質の抗原結合に関る可変領域(VHとVLドメイン)は、ほとんど共通したアミノ酸配列と立体構造をもつ枠組み構造(フレーム)部分と、抗原認識能を直接担っている6ヶの相補性決定部位(CDR(Complementarity
determining Region))と呼ばれるループ状の部分とからなっている。CDRは、その領域のアミノ酸配列の変化が大きいが、10年ほど以前にChothiaらは、CDR-H3を除く5つのCDRループ構造は、それほど多様なわけではなく、それぞれ、アミノ酸配列と複数の典型的なループ構造との対応関係があることを示した。しかし、V-D-J
リコンビネーション機構によってさまざまな長さとアミノ酸配列が形成されるCDR-H3では、その配列と構造があまりに多様なため、帰納的な「法則」による両者の関係の理解には、本質的に限界がある。抗原結合によって立体構造に変化がおきる場合もあり、同一のアミノ酸配列が複数の構造に対応する場合には、経験的な「法則」探しでは自ら限界があり、コンピュータ・シミュレーションによる演繹的方法を用いる必要がある。
3.3.3.機能予測
構造ゲノム科学では、決定した立体構造に基づいて、その蛋白質の生化学的機能の予測を試みる。しかし、例えば酵素蛋白質の場合、蛋白質分子と酵素反応は常に1対1の対応ではない。一つの蛋白質で2つの機能を持つ場合がある一方で、2つの遺伝子による2つの蛋白質が会合して1つの機能を持つ蛋白質複合体となることもある。どちらかと言うとFunctional
divergenceよりもConvergent evolutionが好まれているようで、平均して一つのフォールド当り1.8の異なる酵素機能が割り当てられており、逆に一つの酵素機能当り2.5ヶのフォールドが割り当てられているという見積もりもある。中でも、あるフォールド(TIM-Barrel,
Alpha-beta hydrolase fold, Rossman fodl,
P-loopcontaining NTP hydrolase fold, ferredoxin
fold等)だけが極めて多くの異なる機能をもつことが知られている。
ところで、現在では、ホモロジーの無い蛋白質を新たに決定すると約20%のドメインのみが新しいフォールドであり、80%は既知のフォールドである。既知のフォールドが持つ生化学機能と同様の機能を持つ確率は約50%ほどとも見積もられているため、立体構造が決定されることによって、その機能が類推される確率は高い。実際、最近の米国U.
C. BerkeleyのS. H. kimらは、ゲノム情報から特定された古細菌Methanococcus
jannaschiiの蛋白質を発現し構造を決定したところ、構造の一部に核酸結合構造と類似の部分構造を見いだし、実際にATP,
GTPの加水反応機能があることを実験的に確認することができている。
それでも、残る50%については、立体構造から分子機能を推定するのは困難である。バイオインフォーマティクスによるアプローチとしては、活性部位を構成する機能モチーフの原子レベルでの構造類似性の解析や、分子表面構造と物性の類似性解析があり、現在の構造ゲノム科学の大きな課題となっている。
![]()
4.理論の役割
4.1.特定の系の立体構造情報に基づく機能発現機構の解析
DNAの二重らせん構造の解明は、直ちにDNAが遺伝情報を保持する仕組みの概要を明らかにした。生体高分子の立体構造が機能解明に結びついた最も輝かしい古典的な例である。しかし、DNAは情報の静的な保持のみでなく、書き込み、読み出し等の機能も持たなければならない。このような機能を立体構造に基づいて理解するためには、二重らせん構造だけでは不十分で、二重らせん構造が環境に応じて形を変える柔らかさに関する知見も必要となる。
生体高分子の立体構造に関して実験的に得られる情報は、しばしば、X線結晶解析見られるように空間解像能が良い場合には時間解像能が乏しく、分光学的手段に見るように時間解像能がよい場合には空間解像能が乏しい。ところが、生体高分子の機能発現の仕組みを理解するためには、時間的にも空間的にもよい解像能の情報を必要とする場合が多い。計算機の中に生体高分子系を構築し、その振る舞いをシミュレートする技法は、計算機の能力の向上、計算技術の進歩の結果、実験的に得られる情報を内挿的に補って、かなり信頼の置ける時間・空間解像能情報を与えるところまできている。この計算的手法は、酵素タンパク質、イオンチャネルタンパク質、電子伝達タンパク質等々の機能発現の研究のための不可欠の手段になりつつある。計算機の能力向上と共に、ごく近い将来幾つかのタンパク質の複合体の発揮する機能の解析にとっても、不可欠な研究手段となるであろう。
タンパク質の機能は多様であるために、その機能発現の仕組みの研究は今のところ、手段として計算機シミュレーションが共通に用いられる点を除けば、それぞれ個別的である印象である。しかし、何れの場合にも、仕組みの深い理解のためには理論と実験の共同作業が不可欠である。
4.2.立体構造の博物学的研究
立体構造情報の量も最近は、配列情報と同様に急速に増えつつある。それにともない、配列解析で行われてきたのにほぼ対応する解析、特定の機能と関係のある部分立体構造の検索、異なる立体構造中に存在する共通部分構造の探索、等が立体構造に関してもなされるようになってきた。立体構造はアミノ酸配列よりも進化過程でよりよく保存されていることが判っているので、立体構造の比較によってより遠縁の蛋白質の間の関係を見出すことができるものと期待される。立体構造の観点からすべての蛋白質を分類しようとする試みも始まっている。その様な試みの結果、蛋白質の立体構造の基本型はせいぜい数千種程度しかないとの見方も生まれつつある。
配列解析の場合、対象は離散的な値しかとらない一次元文字列であるのに対して、立体構造解析では対象が連続量であることを反映して、解析は技術的にも原理的にも格段に複雑である。この分野は現在まさに発展途上にある。
4.3.立体構造予測法の開発・機能予測
現在強力に推進されているいろいろなゲノムプロジェクト等の結果、塩基配列情報は急速に蓄積しつつある。その結果得られる蛋白質のアミノ酸配列情報には、現在のところ立体構造や機能が未知の蛋白質に関するものが多い。そこで配列情報から蛋白質の立体
構造や機能を推定すると言う切実な問題が生じる。配列解析が解決策となる場合もある。立体構造既知の蛋白質にアミノ酸配列が似ていれば立体構造も似ていると推定することができる。しかし、これは二つの蛋白質がかなり近縁である場合にしかうまく行かない。特定の機能に特徴的な部分配列が見出される場合には、それによって機能が推定できる場合もある。ただし、このような方法で機能が推定できる場合はむしろ例外で、一般的には配列と機能の関係はより複雑で、配列から機能を推定することは簡単ではない。本格
的な機能予想には、やはり立体構造情報が必要と思われる。
立体構造予測法開発の試みの一つに、第一原理法と呼ばれるものがある。これは蛋白質の立体構造が自由エネルギー最小の条件で定まっているとするアンフィンゼンのドグマに基づき、博物学的知識を用いることなく、求めようとするものである。この戦略を実行するためには二つの問題を解決する必要がある。第一は、与えられた蛋白質分子がとりうる様々な立体構造の関数として自由エネルギーをできるだけ精密に表現することである。第二は、その自由エネルギーの値が最小となる立体構造を探しあてることである。これは非常に高次元の立体構造空間に定義された自由エネルギー曲面を上手に歩いて、エネルー値が最小の点に到達する問題である。第二の最小点を求める問題の難易度は、もちろん自由エネルギー曲面がどの程度おとなしい関数であるかによって決まるわけだが、実はこれが極めて荒々しい地形を持っていることが刊っており、その様な地形上の最小点を求めることを目指した数々の数学的技術が過去に試されてきているが、成功の見通しが立っているとは言いがたい。
第一原理に基づく立体構造予測法の見通しが立たないこともあって、博物学的知見を援用する予測法開発の研究も活発である。上述のように立体構造解析等の結果、蛋白質立体構造の基本型はせいぜい数千種程度との推定がなされている。これが正しければ、立体構造予測は、無限の可能性のうちの一つを予測するのではなく、有限の数千種のうちの一つを予測することになるので、問題は随分と簡単になることが期待される。この期待に基づくのが、博物学的予測法でその実用化への期待が高い。この方法の基礎の一つは、蛋白質立体構造を分類し、その基本型を同定することだが、一見単純に見えるこの問題も、似ているとは何がどのように似ているのか、似ているとされる場合に、その背景にあるのは生物進化的理由かあるいは物理化学的理由か等を追求していくと、極めて複雑で高度な問題へと発展していく。
4.4.新しい実験手段開発への寄与
生体高分子の立体構造の実験的決定が急速に進むようになったのには、主に二つの理由がある。第一は、構造決定に用いられる実験手段−−X線結晶解析、NMR法、電子顕微鏡法、等−−が進歩したこと、第二は、遺伝子操作技術を用いて資料を大量に調整することが可能となったことである。構造生物学の時代に先立って、分子生物学の時代には、遺伝情報に基づく生物学が爆発的に展開したが、そこで用いられる実験手段は化学的色彩の強い比較的小規模なものであった。ところが生体高分子の原子分解能立体構造決定に用いられる実験手段はいずれも、より大規模で物理的色彩の強いものとなってきた。しかも、測定法の面でも、測定によって選られたデータの解析法の面でも、高度な理論的裏付けを必要とするようになってきた。さらに、high
through-put実験への強い需要等に押されて、実験手段は今後とも多様な進歩が予想される。今後のわが国における構造生物学の発展を豊かなものとするために、新しい技術の展開をも自前で進める覚悟が必要である。その際、理論家との協力は不可欠である。
![]()
5.構造生物学の主要な課題
5.1.重要な生命現象を担うマシナリーの構造決定と機能発現機構の解明
−−立体構造と機能理解との間の距離はいろいろ−−
生命活動は細胞という単位を基本として営まれる。その中で、蛋白質や核酸などの巨大分子からなる機械装置が様々な仕事を分担し、エネルギー、信号、物質の受け渡しをする複雑で巨大なネットワークを構成している。特定の仕事には必ず特定の分子機械があり、その種類は数万を越える。それぞれの分子機械のきわめて巧妙な働きは、部品として働く個々の原子の立体配置とその柔軟性、機動性、および反応活性によって規定される。
構造生物学の主要な課題は、これらの分子機械の動作メカニズムを分子機械を構成する原子の立体配置に基づき解明し、さらには様々な分子機械の細胞内外での立体配置と相互作用の動態を見ることにより、ダイナミックな分子機械ネットワークの動きと動作機構を解明することにある。言い換えれば、生命現象を原子レベルから細胞レベルでの3次元動画として見せることにある。生命現象の究極的理解はこのステップを踏まずして実現不可能であり、原子レベルでの理解はその最も基盤をなすものとして重要である。生命科学における構造生物学の重要性は、そのように認識されるべきである。
生命現象のなかでも最も基幹となる部分、例えば遺伝子発現および複製、蛋白質合成、信号伝達処理、物質代謝、エネルギー代謝、物質輸送などは、あらゆる生物あらゆる細胞に共通な機構であり、これらの活動を支える分子機械群の個々の立体構造と相互作用を原子レベル細胞レベルで明らかにすることが、構造生物学の最重要課題である。また、これらの基幹部分から派生して進化したと思われる高次機能についても、例えば運動、感覚から脳機能に至るまで、生命特有の興味深い特徴、すなわち、非常に緻密で精巧である一方、柔軟で曖昧な部分を含む動作機構に支えられており、こういった特徴の裏にある物理的背景を明らかにし、生命現象を包括的に理解する基盤を与えることが、構造生物学のもう一つの主要課題である。こういった側面は、生命現象を支える役者(遺伝子)を同定するゲノム科学には期待できない部分であり、ゲノム科学から提供される情報の上に構造生物学の成果のみが構築展開できる、生命現象の理解にとって必須な情報体系である。
ただし、現在の構造生物学が生命科学に提供できる情報には、その内容の深さと豊かさにおいて対象とする系ごとに差がある。分子機械の立体構造からその動作機構が明白な場合もあるが、必ずしもそういったケースばかりではない。例えば筋細胞中で酸素分子貯蔵の役割を果たすミオグロビン、筋収縮制御に関わるカルシウムイオンを貯蔵するCa結合タンパク等は、その立体構造から動作機構の推定が容易である。分子間の認識と結合特異性は、原子の立体配置から説明が容易である場合が多い。より複雑な機能の場合でも、構造からその分子の動態が予測される場合もある。最近の最も典型的な例は、英国の研究機関から発表されたF1ATP合成酵素の立体構造である。ATP合成を担うサブユニットの3回回転対称構造からの部分的変異から、それらのサブユニットの中心軸にある偏心した軸構造をなす別のサブユニットの回転によるATP合成機構が推察され、その後我が国の研究グループにより逆反応(ATP加水分解)時の回転が光学顕微鏡を用いて観察された。一方で、例えば光合成に関わる光反応中心複合体の場合は、10数年前に膜蛋白質として初めて立体構造が解明され電子伝達経路が明らかにされたものの、電子伝達という複雑で膨大な量子計算を必要とする機能ゆえに、完全な動作機構の理解には至っていない。以下にそのような具体例をさらに詳しく例示する。
5.1.1.立体構造が直ちに機能発現の仕組みを示唆する場合
蛋白質の立体構造、特に機能発現を行う相手のリガンド分子との複合体立体構造が決定された場合には、その立体構造から、機能発現の仕組みが推定される場合が多い。具体的な例として、DNA修復酵素の機能発現の例を示す。
紫外線や化学物質によってDNAに損傷がおこり、突然変異を引き起こして細胞死から生物個体の死にまで至るが、生物はこのような損傷を自身で修復する機能を持っている。蛋白工研の森川らは、cis-synピリミジン・ダイマーのグリコシル結合を加水分解して取り除く反応を触媒する、バクテリオファージT4由来のエンドヌクレアーゼVの構造決定を行った。既に1990年に基質フリー状態の立体構造を決定していたが、その構造とアミノ酸置換の実験から活性部位は推定されたものの、エンドヌクレアーゼVがピリミジン・ダイマーをどのようにして特異的に認識しているかは理解できなかった。変異した塩基はDNA二重鎖の内側に入っており、二重鎖の外側からの認識はDNA鎖の折れ曲がりしか考えられず、直接的な認識機構についての手がかりがどうしても得られなかったのである。
ところが、その5年後の1995年に、cis-synチミン・ダイマーを含む12塩基対のDNA二重鎖との複合体結晶構造の決定に成功したところ、驚いたことにチミン・ダイマーの5'側チミンに相補的なアデミン塩基が、DNAらせん構造の外側へ大きくフリッピング・アウトしていた。エンドヌクレアーゼVの構造はフリーな時とほとんど同一であり、このフリッピング・アウトしたアデニン塩基を蛋白質表面のポケットが相補的に捉えていたのである。逆に、チミン・ダイマーの塩基自体は蛋白質には直接認識されていなかった。このアデニンのフリッピング・アウトと周囲の構造変化によってDNAの二重鎖もほぐれていたが、その二重鎖中の穴の中に、アミノ酸置換によって推定されていた活性部位の残基が入り込んでいた。
このように、紫外線の損傷によって形成されたピリミジン・ダイマー構造が、それを修復する酵素によってどのように認識されるかという機能発現のメカニズムが、ピリミジン・ダイマーを相補する側の塩基のフリッピング・アウト構造の認識による、ということが理解されたのである。このメカニズムは、この研究以後、DNA修復酵素の損傷塩基認識機構の一般的な一つとして考えられるようになった。
5.1.2.多くの条件下での立体構造決定が必要な場合
呼吸酵素であるチトクロム酸化酵素は、プロトンの能動輸送によって酸素還元によって得られる膨大なエネルギーを細胞が利用可能なエネルギーに変換している。実際には、1分子の酸素を還元するために膜の内側から4個のプロトンを汲上げて消費すると同時に、4個のプロトンを膜の内側から外側へ汲上げる。その結果、プロトンの数で12個分の濃度勾配を形成することになる。このメカニズムを解明するため、酸素還元によって作られる自由エネルギーが一挙に放出することなく、幾つかのステップに分けてプロトンを一つずつ輸送し、効率の良いエネルギー変換を行っている可能性を考慮して、各段階の構造決定が進められている。蛋白質の各反応中間体の結晶構造解析によって蛋白質のなかで営まれている物理化学を明らかにし、呼吸によるエネルギー変換機構についての理解を深めようとする戦略である。従って、構造解析の精度も物理化学を語るに十分な精度でなくてはならず、ペプチド鎖骨格のリボン図がかける程度の精度では役に立たない。多くの場合、蛋白質内では、高い規則性をもって段階的に進行する化学によって高度に制御された機能をもたらしている。この段階的に進行する化学を理解するための構造解析は、克服しなければならない困難な問題を多く抱えているが、これらはX線結晶構造解析の例外的な課題ではなく、むしろ中心的な課題になろう。
5.1.3.深い理論的解析を必要とする場合
生体高分子はあくまで分子であるから、量子力学・統計力学等で記述されるミクロな世界の法則にしたがってその機能を発揮する。従って、マクロな世界で経験的に培われたわれわれの直感のみでは、その立体構造から機能発現の仕組みを読み取ることが困難な場合が多い。5.1.1および5.1.2で述べたようにようなケースはむしろ例外的なのかも知れない。もちろんそのような場合においても、機能発現における個々のアミノ酸残基の役割を問うようなレベルになると素朴な直感は無力になる。ミクロな世界の法則で理論武装したしっかりした解析を必要とする。その典型例としてcytochrome
cの例を挙げよう。この蛋白質は細胞膜表面に存在し、呼吸鎖中の重要な電子伝達反応を触媒する。この分子のX線結晶構造は、カツオ心筋由来の分子について、1973年大阪大学蛋白質研究所の角戸研究室により解析された。立体構造は解明されたが、本来量子力学的な現象である電子伝達反応の詳細をその立体構造から直感的に推察することは全くできなかった。しかし、この構造の解明は、タンパク質分子中を電子がいかに移動するかを量子力学的に解明しようとする研究や、電子が移動できるミクロな条件が熱揺らぎによって満たされる過程の統計力学的研究の流れを作り出した。このような研究は生物学と言うよりはむしろ化学の世界の問題と捉えられていることが多いが、大きな研究の分野を開くきっかけを作った研究であった。このような研究の例は、蛋白質の多様性を反映して多様にして豊富である。その様な多くの系の深い理解の上に、物理学・化学等の物質科学と生命科学とが融合した新しいパラダイムが生まれてくるものと期待される。
5.2.蛋白質全ファミリーの立体構造決定プロジェクト
5.2.1.意義と可能性
生体分子の立体構造に基づいて生命現象を理解する「構造生物学」の研究は、大きく分けて3つの過程からなる。まず各蛋白質の立体構造を決定し、次に立体構造に基づいて分子機能を解析し、最終的にそれらを総合して各蛋白質の生物学的役割を理解する。
現在の分子生物学は多くの場合、分子単位、分子レベルで理解されており、その分子を構成する原子の立体配置に基づいた分子機能の理解には程遠い。その理由は、数多くの生命現象を枚挙しつつある分子生物学の研究分野と、時間をかけて一つ一つの蛋白質の立体構造解析を行う研究分野との間にかなりの隔たりがあり、これらの研究分野の間に分子機能解析という大きな研究分野が横たわっているためである。分子機能解析の分野はまだまだ発展途上にあるが、幸い立体構造解析の分野ではX線結晶解析やNMRなどの実験手法もある程度確立し、生体高分子の立体構造を決定するために必要な予算や時間についてもある程度目処がつく時代になった。そこでまず、多くの蛋白質の立体構造を解析し、その立体構造構築原理を明らかにして次世代の分子機能解析に備えようというのが、蛋白質全ファミリーの立体構造決定プロジェクトである。
蛋白質の立体構造は、ドメインと呼ばれる約100−200個のアミノ酸残基の構造単位がいくつか集まってできている。各ドメインのペプチド骨格の折れ畳み方(フォールド)には似たものがたくさんあり、明らかに異なるものを独立なフォールドとしてクラス分けすることができると考えられている。つまり、フォールドは約千種類の立体構造ファミリーに分けられると推定されている。化学物質が約百種類の元素からできているように、蛋白質の立体構造は千種類の立体構造ファミリーの組み合わせによってできていると考える訳である。このうち約60%の立体構造ファミリーがこれまでに解明されている。残された約40%の立体構造ファミリーのフォールドを解明するために、世界中が協力してなるべく重複の無い約1万個の蛋白質を選び、それらの立体構造をこの数年間で解析するプロジェクトが計画されている。これによって約千種類ある全フォールドの立体構造構築原理が解明できれば、ゲノムプロジェクトで大量に解明されつつある蛋白質のアミノ酸配列から、即座に立体構造を予測することも原理的には可能になる。そうなれば立体構造をもとにして分子機能解析へ進む研究が加速され、現在の「分子生物学」は、いわば「原子生物学」と言っても良い本来の「立体構造分子生物学」へと展開し、生命科学は一大転機を迎えることになる。
5.2.2.生命科学への波及効果
ヒトを始めとする真核生物の蛋白質は、不安定であるか、あるいは結晶化が困難であるなどの理由により、立体構造決定が難しいことが多い。ヒトゲノム解析は2003年に完了しポストゲノム時代を迎えるが、その際、機能予測と解析に重要な立体構造に関しては、実験的解析が難しいものについては予測に頼らざるを得ない。その時点で約千種類ある全フォールドの立体構造構築原理があらかじめ解明できていれば、ゲノムプロジェクトで大量に解明されつつある蛋白質のアミノ酸配列から即座に立体構造を予測できる他、人工機能蛋白質を作ることも原理的には可能となる。また、ある蛋白質が創薬のターゲットとなるような場合、たとえその蛋白質の立体構造解析ができなくとも、直ちに創薬研究に着手することがある程度可能となるし、より厳密で論理的な薬分子設計のためにその蛋白質自身の精密立体構造が必要な場合には、予測構造を基にした分子置換法で結晶構造解析を行うことにより、実験的構造解明を短時間で完了することもできる。
約千種類ある全フォールドの立体構造構築原理が解明でき、数多くの蛋白質の立体構造が決定されれば、それをきっかけとして蛋白質機能解析法も飛躍的に発展することが期待される。そのためには、様々な分子機能解析法に習熟し、各解析法の長所・欠点を幅広く理解し、必要に応じて新規な分子機能解析法の開発する能力のある学際領域の人材育成が切望される。この分子機能解析法の進展によって、真の「構造生物学」へさらに一歩近づくことが可能となる。
5.2.3.世界の動きと我が国での対応
約千種類の基本フォールドのうち残り約40%とされる基本フォールドを解明するために、約1万個の蛋白質の立体構造を主にX線結晶解析で決定するプロジェクトが、米国を中心に、ヨーロッパなどでも始まりつつある。とくに米国では2000年秋に3〜6のグループを選び、その後5年間をかけて、予算100億円以上の規模で、蛋白質の全フォールド決定に向けた解析作業をすることになっている。そこで得られたデータは、ゲノム解析と類似の方法で直ちに公開され、世界中の研究者が利用できるようになる。日本の現状は、立体構造解析のできる人材が少なく、また解析を大々的に行える組織も無いため、欧米のように短期間に多数の立体構造解析を行うことは難しい。かといって欧米のデータを利用するだけでは、日本に対する「ただ乗り論」が持ち出される懸念がある。
このような欧米の動きに対して日本では、NMRで全フォールド決定を目指すプロジェクトが理研ゲノムサイエンスセンター(GSC)で始められつつある。ここでターゲットとされるのは、現在解析が進行中のマウスcDNAから発現される蛋白質群であるが、国際的に計画されている全フォールド決定のための国際協力プロジェクトの一環としても進められる。一方、理研播磨SPring8では、その世界で最も強力で高輝度な放射光X線を利用して、X線結晶解析による構造決定プロジェクトが始められている。ただし、このプロジェクトの基本的目的は、上述のような基本フォールドをリストアップすることではなく、ある高度好熱菌細胞内の生体分子の立体構造をすべて決定して、生体分子の分子機能を物理化学的なレベルで解析し、それらに基づいて一つの細胞内での生命現象を包括的にかつ原子レベルの分解能で理解しようとするものである。つまりこれは、次世代の基本的生命科学を志向した壮大な研究のごく初期過程に過ぎない。このように将来の生物学を見据えた構造生物学的プロジェクトは、世界的にみても今のところ日本のこのプロジェクトのみであるが、そのターゲットの壮大さゆえに、あまり理解が得られていないのが現状である。もちろんその研究成果は、基本フォールド全解明プロジェクトにも貢献することは明らかである。
構造ゲノム科学に関するプロジェクトが国内に2つしかなく、しかもこの分野の人材が少ない日本の現状が続けば、この数年間に新規な基本フォールドを決定することで日本が世界に貢献できる割合は、多くても1割以下であろう。
資料:
Structural Genomics Projects in the United
States
・UCLA/LANL(D. Eisenberg et al.) Pyrobaculum
aerophilum-Proteins(24%) of medical relevance
identified by homology with proteins in NCBI's
OMlM.
・UC Berkley(Sung-Ho Kim et al.) The hyperthermophilic
archaeon Methanococcus jannaschii proteins
by X-ray.
・BNL/Rockefeller Univ.(Bill Studier et al.)
Focused on Yeast proteins.
・Rutgers University(Guy Montelione et al.)
Emphasis on NMR;Targets of relevance to human
disease;broad conserved metazoan genes;human
pathogens;From a set of homologous proteins
from different species.
・Northwestern University(Paul Bash et al.)
To obtain a "basis set" of protein
folds;Structures with a wide phylogenetic
distribution.
・CARB/TIGR(John Moult I) Structures for
the unannotated open reading frames from
Haemophilus influenzae.
Structural Genomics Projects in Europe
・Protein Structure Factory(Heinemann of
Max Delbrueck Center et al.) High throughput
structure analysis of medically relevant
proteins. A consortium of groups funded by
the German Ministry for Research and Technology(BMBF)
with approx. $20 million over 5 years. At
least l00 structures of thermostable proteins/antibiotic
targets/ translation components.
・Structural Biology Industrial Platform(SBIP)
(EU) Consortium of l5 companies including
some of Europe's largest pharmaceutical industries.
Structural Genomics Projects in Japan
・RIKEN/Tokyo University/(S. Yokoyama et
al.) Mouse cDNA; domain fold determination
by NMR
Structural and Functional Genomics in Japan
・Osaka University/RIKEN/(S. Kuramitsu et
al.)高度好熱菌 丸ごと一匹プロジェクト−基本的生命現象の系統的解明−Thermus
thermophilus HB8; structural and functional
whole cell project, (since l995); structural
genomics (X-ray crystallography (or NMR));functional
analyses. (reaction kinetics, molecular biology,
unknown genes, tips, proteome) → "atomic
biology"
5.2.4.問題点
構造生物学のドグマとして考えられている配列−構造−機能の間の相関は、実はそれほど強いものではないという点が、蛋白質の全ファミリー立体構造決定プロジェクトにおける基本的な問題点となっている。すなわち、この相関の弱さのために、機能未知のタンパク質の立体構造を決定しても直ちに機能を特定することは容易でなく、プロジェクトの成果からは直接、各遺伝子産物の機能解析に結びつかないのではないかという疑問が提示されている。
実際、タンパク質の生化学的機能が類似していても、立体構造まで類似しているとは限らず、また立体構造もアミノ酸配列も類似していても、タンパク質としての機能が同一とは限らない。
機能と立体構造との関係は、以前は単純に考えられていて、ある構造単位は特定の機能とのみ対応していると思われていた。例えば、Zn-フィンガー・モチーフやロイシンジッパー・モチーフがあれば、DNA結合部位だろうと推定された。しかし、今や多くの反例が登場してしまい、DNA結合機能とは無関係なタンパク質中にも、Zn-フィンガーやロイシンジッパーが発見されている。同一の機能を持ちながら、立体構造が異なる例として有名なものには、セリン・プロテアーゼのトリプシンとズブチリシンがあり、活性部位のアミノ酸側鎖の配置は極めて良く似ているにもかかわらず、全体のフォールドは全く異なる。
以上のように、蛋白質分子としては全く別々の祖先から同じような機能を得るために似た構造をもつに至った例や、配列に比べて構造が保存されやすいとはいうものの機能に関係しない部分は大きく構造が変わってしまった例もある。このため、蛋白質の全体構造が決定されても、その情報のみから機能を類推するのは自明のことではない。
さらに、タンパク質の生化学的機能が類似していても、それは生物学的機能とは別の問題である。例えばバクテリアのペリプラズムにある低分子リガンド結合タンパク質は、転写調節因子として働くDNA結合タンパク質の中のある一つの構造単位(ドメイン)を構成しており、調節分子結合ドメインとして利用される。また、類似の構造は、膜タンパク質であるグルタミン酸受容体のアゴニスト結合ドメインとしても利用されている。このドメインの機能は、広い意味では低分子結合として同一ではあるが、生命個体における機能としてはそれぞれ全く異なっているわけである。
この問題点に焦点を当てた研究が現在では盛んになされており、分子認識を行ったり酵素反応をおこなう活性部位の詳細な構造やその表面構造の幾何学的性質だけでなく、物理化学的性質、進化的性質に焦点を絞った研究や、配列と構造に対して同時にマルチプル・アラインメントを行う研究などが行われている。
また、ゲノム情報の理解のためには立体構造決定のみでアプローチする必要はなく、機能ゲノム科学(functional
genomics)による対象蛋白質の生物学的機能の解析結果も積極的に取り込む必要があろう。構造ゲノム科学と機能ゲノム科学はゲノム科学という広い学問体系の車輪の両輪であり、それにかかわる発生生物学、分子生物学、構造生物学、バイオインフォーマティクスなど、さまざまな学問の成果によって遺伝子型から表現型への橋渡しが何本もかかることこそが、ゲノム科学を成熟させることになると思われる。
全く別の技術的な問題として、このプロジェクトを推進するにあたってはハイ−スループット(高速・高効率)で構造決定をする解析技術が必須である点が挙げられる。全世界の研究室が協力し、研究資源をそのためだけに投資したとしても、数年以内に約1万個の蛋白質の構造解析を完了することは容易なことではない。X線結晶解析法やNMR法に関してはデータ収集法と解析法が成熟しつつあるので、今後の測定手法の更なる合理化によってある程度ハイ−スループット化が期待できる。それにしても、そのための技術開発にまず真剣に取り組むことが必要であろう。また、全プロテオームの20−35%を占める膜蛋白質については構造決定のための技術がいまだ確立されておらず、2次元、3次元の結晶化技術や、電子顕微鏡による単粒子解析等、まだ現在研究段階にある。今後は、従来の方法をより高度化すると共に、膜蛋白質の構造決定に対する取り組みを重点化する必要がある。膜蛋白質の構造解析を含めた迅速構造解析法の開発と確立にこそ、限られた研究資源を集中すべき時期であろうと思われる。
もし10,000個の蛋白質の立体構造を解析した結果、あらゆる蛋白質の立体構造を予測できるようになるなら、べータバレル蛋白質を始めとして頻繁に出現する立体構造のファミリーについては、これまでの知識からだけでも予測ができて良いように思われる。もしそれが不可能なら、あと10,000個あるいは100,000個の立体構造を解析しても、立体構造予測は難しいように思われる。予測が目的なら、自然界に現存する蛋白質を数多く解析するだけではなく、変異型蛋白質を含む種々の人工蛋白質についても解析するなど、幅広いアプローチが必要になるかも知れない。
5.3.新しい実験的立体構造決定法の開発
5.3.1.より大きなより複雑な系
細胞という単位を基本として営まれる生命活動では、様々な分子機械が様々な仕事を分担し、エネルギー、信号、物質の受け渡しをする複雑で巨大なネットワークを形成しており、そのネットワークの活動そのものが生命現象であるとも言える。つまり、個々の分子機械の構造と同様に、分子機械間の相互作用を原子レベルから細胞レベルにいたるまで様々な階層で解明しない限り、生命現象を十分に理解することはできない。相互作用は分子種によって様々な時間空間スケールで行われ、必要に応じて会合解離を繰り返す場合もあり、複数種の複数分子によって構成される巨大な複合体を安定に形成する場合もある。
各サブユニットは分子量数万Daでも、複合体の分子量は百万から一千万Da以上になる場合もあり、ウィルスなどはその極端な例と見なすことができる。繊維状会合体では長さが数十ミクロンを越え、構成サブユニット数が数万個を越える場合も珍しくない。
単分子の場合はもちろん、複合体でも構成分子数が比較的少なくコンパクトで安定な形状の場合は結晶化が可能な場合が多く、極低温での回折データ収集法と単色高輝度の放射光X線を使うことにより、X線結晶解析法がその威力を発揮して高分解能の立体構造を見せてくれる。例えば分子シャペロンGroEL-GroES複合体は、2種めサブユニットそれぞれの7量体が会合した複合体の両極性2量体、つまり総サブユニット数28の大きな複合体であるが、X線結晶構造解析の結果、複合体中に形成される空洞にミスフォールドした蛋白質をトラップし、ほどいてリリースする過程で正しいフォールディングを助ける仕組みがそれ自身の大きな構造変化とともに明らかにされた。タンバク質合成装置であるリボソームは数種類のRNAと数十種類の蛋白質の巨大な複合体であり、全く対称性を持たないため構造解析は困難を極めたが、それでも最近良質な結晶が得られたために結晶構造解析が高分解能へ向けて進められている。
しかし、どうしても結晶化できない場合にはどのような可能性があるか? あるいは、結晶中では困難な複合体の機能的動態観察はどのように行うか? こういった場合には電子顕微鏡がその能力を十分に発揮してくれるであろう。最近の電子顕微鏡法の発展は、装置と解析法、つまりハードとソフトの両面においてめざましい。試料の急速凍結氷包埋法と極低温像記録法によれば、陰染色試料を室温で記録するのに比べ像質と到達分解能に飛躍的改善が得られる。電解放射型電子銃の干渉性と輝度の高い電子線の導入も像質と分解能向上に大きな貢献をした。対象となる複合体粒子の2次元結晶が得られれば、様々な傾斜角で投影像を記録し解析することで、原子モデルの構築が可能な分解能(3A程度)が、数例ではあるがすでに達成されている。また、溶液中に浮遊する複合体粒子でも、多数の像を記録し面内で整列させた後、電子ビームに対する向きごとに分別した平均像を逆投影することにより粒子の立体像を再構成することが可能である。この場合、分解能は解析に用いられた粒子像の数に依存して向上し、原理的には蛋白質の主鎖や側鎖を認識できる分解能も得られるはずであるが、そのためには百万個を越える粒子像が必要であると言われている。現在のところ、例えばリボソームでは数万個の粒子像から6〜7A分解能が得られている。主鎖側鎖の認識を可能にする3A分解能にはまだ隔たりがあるが、様々な反応中間体の構造を次々と解析できるため、それらを連結してタンパク質合成の立体動画が作られるまでになった。分解能で劣るとはいえ、これはX線結晶学には簡単にまねのできない電子顕微鏡の特異な能力である。そして、電子顕微鏡法の発展はまだとどまるところを知らず、ハード・ソフトの両面においてさらなる飛躍が期待されており、原子分解能が達成されるのもそう遠い未来の話ではないと期待される。
5.3.2.揺らぎやダイナミックス情報をも得られる高精度構造決定
5.1で述べたように、立体構造と機能理解との間の距離はいろいろである。多くの場合、X線結晶解析やNMRで得られる平均構造情報のみでは、機能発現の仕組みを深く理解するのは不十分である。それを補う一つの方法が理論的な計算機シミュレーション法を用いることであるが、揺らぎやダイナミックスに関する詳細情報をより直接に実験測定により得る方法を開発することが強く望まれる。X線結晶解析法においてもNMR法においても、測定法と解析法を共に現在よりもさらに高精度化することにより、揺らぎやダイナミックスに関して得られる情報の質を格段に高上し得る余地は十分にある。構造生物学の一つの進むべき方向は、機能発現機構の深い理解を目指した高精度構造解析法の開発である。
5.3.3.時間軸測定
構造生物学の分野では、新規な生体高分子の立体構造決定に加えて、生体機能の作用機序を究明することの重要性がますます高まっている。しかし、反応中の生体高分子が構造を変化させて基質を認識したり反応を触媒したりする様子は、現在でもまだ多くの傍証によって推察することが一般的である。従って、生体高分子の機能を直接的に明らかにし精確に理解するには、従来の3次元構造に時間軸を導入した、4次元での動的構造解析が必要である。
X線結晶構造解析で得られる電子密度図は、結晶中の多数の分子の空間的、時間的な平均である。従来はX線回折強度測定に長時間を要したため、酵素等の反応中間体の構造解析は不可能であった。しかし、放射光の出現や各種のX線検出器の進歩により、数分からナノ秒に至る時間領域で精密な動的構造を直接観測することが現実的なものとなった。1990年にSchlichtingによってHa-Ras-p21タンパク質中に結合している加水分解直前のGTPが白色ラウエ法で捕らえられて以来、ヨーロッパ放射光施設ESRFを中心に約10例の動的構造解析がなされ、その多くがNature、Science誌に掲載されている。我が国においても、科学研究費補助金・重点領域研究「動的蛋白結晶解析」(平成5年〜8年度、領域代表:坂部知平)によって、高エネルギー加速器研究機構・物質構造科学研究所・放射光研究施設PFリングの実験ステーションBL-18Bにおいて白色ラウエ法を利用するミリ秒時分割カメラが開発され、さらに現在は、PF-ARリングにシングルバンチを利用するナノ秒領域の時間分割測定用実験ステーションが建設中である。また、第三世代放射光施設SPring-8にも、白色ラウエ法の可能な実験ステーションBL44B2が建設され運用を開始している。さらに第四世代放射光が実現すれば、より高輝度のパルスX線が利用可能となる。また、この種のデータ収集には、ピクセル・アレイ検出器と呼ばれる高速高感度の大画面2次元検出器に大きな期待が寄せられている。
白色ラウエ法は測定時間をミリ秒以下に短縮できるが、一方で結晶性に対して厳しい制限があることも上記の重点領域研究等によって明らかになった。そのため、白色ラウエ法のような測定時間短縮による動的構造解析のアプローチと相補的な研究方法として、温度・pH・その他の実験条件を調整して特定の反応中間体の寿命を秒から分の時間領域に延ばし、X線回折データ収集時間よりも長寿命の準安定化状態をとらえ、単色X線を用いた従来型の測定方法による時間分割構造解析を行う試みが、最近は多く行われるようになっている。
ただ、結晶構造による構造変化の時間軸測定における基本的問題点は、機能発現にともなう構造変化が大きい場合、それが結晶場に抑えられるか、あるいは逆に結晶構造を破壊するために、構造変化を観測できない場合が存在することである。
5.3.4.迅速構造解析
X線結晶構造解析による蛋白質の構造解析には、時間がかかるというのが一般的な認識である。しかし、蛋白質構造解析法とその周辺分野の発展により、最近の構造解析は非常に迅速に行われるようになってきている。解析のボトルネックは依然として結晶化にあるが、一旦良好な結晶が得られたならば、数ヶ月から数週間、時には数日で構造解析が完了することすら可能になってきている。しかし、ポストゲノム科学時代の構造生物学を展開するためには、解析をさらにスピードアップすることは必然的な要請である。さらなるスピードアップのためには、結晶化法の開発、モデル構築(電子密度解釈)の自動化、構造精密化のルーチン化などが必要とされる。
近年の蛋白質構造解析の迅速化をもたらした要因として以下のものがある。(1)シンクロトロン放射光の普及:非常に高輝度なシンクロトロン放射光を利用することで、これまでは解析不可能であった回折能の低い結晶から回折データが収集できるようになり、解析可能な結晶の制限が緩和された。(2)多波長異常分散法:シンクロトロン放射光の波長選択性を利用することで、多波長異常分散法という新しい解析法が生まれた。特にセレノメチオニンを利用した多波長異常分散法の利用により、重原子同形置換体の調製にかかっていた時間が大幅に短縮された。(3)遺伝子工学の発展:タンパク質の大量調製が可能になったため精製が高速化し、結晶化に必要なスクリーニングが簡単に行えるようになった。また、部位指定突然変異法により重原子結合部位を導入し、構造解析に利用することもはじまった。特に重要なのは、Seの導入部位としてのメチオニンの導入、Hg結合部位としてのシステインの導入などである。(4)計算方法・解析プログラムの発展:計算機の高速化とともに、解析のための新しい方法、ソフトウエアが開発された。重原子パラメータの精密化におけるmaximum
likelihood法と電子密度の改良法は、迅速構造解析に寄与するところが大きかった。(5)回折実験法の進歩:極低温での回折実験法の開発により、単一の結晶から非常に良質の回折データセットを複数回収集する事が可能になった。特に、第三世代の放射光施設においては、高輝度X線による照射損傷を抑えるため、極低温でのデータ収集法は不可欠のものとなっている。(6)検出器の進歩:S/Nの高い良質のデータを短時間に収集するためには、イメージングプレートやCCDなど、2次元検出器の性能向上も大きな貢献をしている。(7)放射光利用により10ミクロン級の微小結晶が解析可能となった。
我が国における様々な技術開発は、シンクロトロン放射光利用技術や、イメージングプレート検出器の高速化、高感度化などの分野で高いレベルの国際的貢献を果たしているが、半導体技術先進国として先進的2次元検出器の開発にさらに大きな貢献ができるポテンシャルを秘めながら、十分にその力を発揮できていないのが現状である。ポストゲノム科学の時代にこそ、この方面での技術開発に予算と人材確保の努力が期待される。
また、迅速構造解析にとっての最大のボトルネックである蛋白の大量精製と結晶化については、特に遺伝子の約30%を占めると言われる膜蛋白質についての対応が遅れており、あらたな分子生物学手法の開発による容易な大量発現システム構築法の確立と、より系統的かつ論理的な結晶化法の早急な開発が望まれ、この分野の人材確保のためにも構造生物学分野のすそ野を広げることが急務であろう。
5.3.5.構造解析ソフトウエアの開発
構造解析のソフトウエアとしては、データ収集・処理に関するもの、データ解析に関するもの、解析結果を利用するためのものに分けることができる。回折データ収集・処理に関するソフトウエアは、各放射光施設において独自のものが開発されてきた。蛋白質からの回折データ収集は、2次元検出器を利用して行うために、自動指数付け、積分強度測定などのプログラムが必要とされる。現在のところ、DENZO、MOSFLMなどが最もよく使われている。蛋白質構造解析関係のソフトウエアとしては、イギリスの結晶学者が中心となって作製したCCP4(Collaborative
Computational Project-Number 4)システムが最も一般的に利用されている。これは構造を得るために必要とされる多くのプログラムからなっており、データに合わせて多数あるプログラムを組み換えたり、パラメータを決めたりすることができるようになっている。解析に精通している人には問題ないが、初心者にはやや使い難いところもある。構造ゲノム科学プロジェクトに関連してつくられたソフトにSolveがある。このプログラムは、回折データから電子密度計算に至る一連の流れを、プログラムに独自の判断機構を持たせて自動的に行うことができるようにつくられている。電子密度から構造を構築するためには、Oというプログラムが最もよく使われている。ARP/wARPというプログラムでは、位相改良の他、電子密度の自動トレース、初期モデルの構築を行うことができる。この種の電子密度自動解釈ソフトは、現在のところ分解能等に制限が多く実用的ではないが、近い将来より優れた使い易いものになると期待される。構造の精密化にはX-PLORが用いられてきた。現在X-PLORは新たなサポートがされておらず、CNSと形を変えて開発がすすめられている。CNSはインターフェイスがHTML形式なのでパラメータの入力等が簡単である。
得られた立体構造は、PDB(Protein Data Bank)が取り決めた形式で、原子座標、温度因子などが登録されるが、PDB形式のデータファイルを読みこみ、グラフィック表示できるソフトウエアとしては、Rasmol、Molscript、GRASPなどがあり、論文の図の作成にもよく使われる。
ただ、残念ながらこれらのソフトウエアはほとんど海外の研究者の手によるもので、国内の研究者や技術者によって開発され全世界で広く使われているものがほとんどないのが現状である。これは、国内での構造生物学分野のすそ野の狭さによるものであり、この境界領域の研究分野で広く人材を確保するためにも、構造生物学を広く普及させ人材層を厚くすることが、国際貢献のためにも急務である。
5.4.細胞生物学への新しい寄与:空間と時間を扱う広義の構造生物学
分子生物学が技術体系として近代生物学の基盤となって以来、分子生物学的手法は、発生生物学、神経生物学、植物生理学などへと直ちに応用された。これらの研究では、細胞内の分子レベルでの研究のみならず、細胞機能レベル、多細胞レベル、器官レベル、個体レベルでの研究と階層的に問題を追求する必要が認識された。この中で、分子生物学的手法に対応する細胞生物学的手法の確立の必要性が自覚されるようになってきた。細胞生物学には、核分裂と細胞周期、輸送、シグナル伝達、細胞内小器官の形成と消滅と言うような、空間での移動と変化の問題が新たに本質的に重要なものとして付け加わる。分子生物学は遺伝子とその発現を根幹とする学問であり、直接時間や空間を意識しないので、あらたに分子、生体高分子集合体、細胞内器官等の空間的分布や時間的変化を正面から取り上げ、生体メカニズムの解明にせまる学問の構築が必要となった。
元来、時間や空間を扱ってきたのは主に物理であった。これらの素養を持ち、かつ生物材料に接していた分野の一つは、蛋白質の構造解明をめざしていた狭義の構造生物学者をふくむ生物物理学者である。これらの人々と、細胞生物学をメカニズム論として構築する立場の人々の間に、広義の構造生物学が誕生した。この中には、シグナル伝達等を原子レベルで解明していく人々だけでなく、原子分解能にこだわらず、もっと大きなレベルでも良いから、あらゆる可能な手段を用いて形と運動とを物理的センスと生物学的センスとで取り扱い、メカニズムを解明していこうという人々が含まれる。
この広義の構造生物学を確立した契機は、Green
Fluorescent Protein(GFP)によるin vivo蛍光ラベル法の開発である。生細胞内で標的とする蛋白質をGFP融合蛋白質とすることにより、細胞内局在や分布の変化を可視化することが出来るようになった。このことは、空間と時間を取り扱う合理的手段の必要性を明らかにした。幸運なことに同時期に、新世代光学顕微鏡の開発、1分子計測とマイクロ操作技術等によるナノバイオロジーの発展があった。ナノバイオロジーは日本から発信された概念(1993)であり、DNA1分子の可視化、ミオシン・アクチンの1分子アッセイとルースカップリング、DNA上の蛋白質のスライディングの証明、F1ATPaseの回転運動の証明など、この分野の主要な成果を国内の研究者がなしたことは特記すべきである。日本が創造性においてリードしている数少ない分野である。
この段階で、構造生物学は第2の発展を遂げつつあると言えよう。関連蛋白質の原子分解能での相互作用解析など、将来にわたって変わらぬ価値を持つ狭義の構造生物学的寄与に加えて、構造生物学は技術的な寄与を細胞生物学に与えている。共焦点顕微鏡、3次元再構成顕微鏡などの新世代光学顕微鏡は、細胞生物学では必須の観測手段として普及している。技術的寄与の第二は、新しいプローブの開発である。GFPを利用したCaセンサーは、すでに脳研究に応用され始めている。今後も構造生物学的に設計された様々な細胞内センサーが開発されるであろう。
広義の構造生物学の概念的な寄与として近未来で確実なものは、生物学への力学的概念の導入であろう。1分子計測をはじめとするナノバイオロジー的手段には、力学的な記述に適したものが含まれる。モーター蛋白質による輸送など、細胞内の時間的変化を伴う現象に対して力学を用いた記述がなされ始めている。このことは、化学を生命現象に導入した生化学の確立以来のエポックであると言えよう。
生命科学を現代科学の一翼として考えた場合、広義の構造生物学はこのように生物学と物理学とを橋渡しする境界領域の一つといえよう。過去には全く存在しなかったものであり、分子生物学に依存した在来技術が飽和しつつある細胞生物学において、新たな技術だけでなく概念的変更も導入する広義の構造生物学は、この意味で脳研究に勝るとも劣らないスケールの現代生物学のフロンティアを形成するポテンシャルを持つと言えよう。
5.5.先端医療・工業等への応用
5.5.1.医療への寄与
ヒトゲノムの配列決定が完了しようとしている現在、ゲノムにコードされた遺伝子と様々な病態との関連性が大きな注目を集めている。構造生物学は、明らかになった疾患遺伝子がどのようなメカニズムで病態を発現するのかを分子レベルで解明することができる重要な方法のひとつである。分子レベルで解明された病態発現メカニズムは、その治療の方策を立てるための重要な手がかりを与える。たとえば、ウイルスの感染によって引き起こされる疾病に対しては、これまでは有効な薬剤が存在せず、ワクチン等の予防措置が唯一の感染を防ぐ手段であった。しかしながら近年ウイルス表面蛋白質の立体構造の解明や細胞への感染機構が解明され、HIVや新型インフルエンザウイルスに対する抗ウイルス剤の開発において重要な役割を演じている。さらにHIVの感染に関わる外皮糖蛋白質gp120とCD4蛋白質複合体の立体構造が解明され、HIVがヒトの免疫機構をどのようにして逃れるのかを知る有力な手がかりが得られている。また、免疫分野における構造生物学の寄与も重要である。MHC蛋白質が、抗原蛋白質の一部である抗原ペプチドを提示する機構の解明は、ペプチド性ワクチンの可能性を提起した。今後の構造生物学は、薬剤の作用点としての膜蛋白質の解析にも重点が置かれていくと考えられる。X線結晶構造解析や電子顕微鏡を用いた膜蛋白質の構造生物学は、我が国の構造生物学が最も得意とする分野の一つである。また大型放射光施設や高分解能NMRの共同利用が推進されれば、構造生物学がますます進展する環境が整うことになる。しかしながら我が国の構造生物学がこれまでにたどってきた歩みは必ずしも満足行くものではなかった。医療分野における構造生物学の重要性の認知は今一つであるといわざるを得ない。海外では、蛋白質の立体構造解析設備を備えた医学・薬学関係の研究室が続々誕生し、Howard
Hughes Medical Instituteをはじめとした民間基金による研究サポート体制も整備され大きな成果をあげている。我々はこれまで医療分野と構造生物学分野の研究交流が充分ではなかったこと反省すべきであると考える。また、医療分野からの期待と実際に構造決定に必要な時間的・資源的な隔たりが大きかったことも問題であろう。両分野の研究交流を効果的に進めるには、蛋白質の発現・結晶化を含めた要素技術の改善とスピードアップが必須であると考える。我々は今、人類の共通の課題である疾病の克服に我が国の科学技術が大きな貢献を期待されていると考える。
5.5.2.創薬への寄与
近年、新しい医薬品の開発において薬物の作用点としての蛋白質分子が注目されている。創薬を行う場合、目的分子が低分子合成化合物であるか、それとも蛋白質なのかによって方策が大きく異なるが、蛋白質に関わる情報が創薬における重要なキーポイントであることは共通している。低分子創薬においては、独自のアッセイ系を構築し、薬物の作用点である蛋白質に対して相互作用する分子を膨大なライブラリの中からスクリーニングすることが主流である。その中で、構造生物学の台頭は、次の点で低分子創薬に寄与しようとしている。そのひとつは、リード化合物の効率の良い最適化である。低分子化合物が作用点である蛋白質と直接相互作用する様子を解明できるので、得られたリード化合物の親和性の向上や薬物動態の改善を効率よく進めることができる。例えば分子上のどの官能基を置換すべきであるのを判断し(ドラッグデザイン)、合成の方策を立てることができる。これはまた有機合成に関する負担を大幅に減少させることができる。実際、1)HIVプロテアーゼ阻害剤、2)インフルエンザウイルス・ノイラミニダーゼ阻害剤の開発において、非常に多くの薬物との複合体構造解析が実施された。構造生物学のもたらす有機合成への負担の軽減は、わが国のように比較的規模の小さい製薬企業にとって、メリットが大きいことを付け加える。また、近年の構造生物学による成長ホルモン(蛋白質性リガンド)の受容体活性化機構の解明は、創薬のターゲットポイントや必要とされる分子形態に関する新しい可能性を生み出した。従来から、蛋白質性の医薬品は経口投与ができず、投与時に患者に大きな負担を与えてきた。構造生物学による受容体活性化機構に関する知見は、蛋白質性リガンドを有機合成された低分子リガンドに置き換えることができる可能性を示した。現在、有力な製薬企業が低分子サイトカインの開発に取り組んでいる。未だ天然の蛋白質性リガンドを上回る薬効を示すには至っていないが、大きな期待を集めている。
一方、構造生物学は蛋白質性医薬品の開発にも重要な知見を与える。蛋白質性医薬品は、高い生物学的活性が期待できるが、蛋白質であるがゆえに抗原性の問題が懸念されてきた。構造生物学によって明らかにされたリガンド−受容体相互作用は、その生理活性を損なわずに抗原性を低下させる方策を与える。また、既存の蛋白質性医薬品を更に高活性化することも可能である。
このように創薬における構造生物学への期待は非常に大きいが、蛋白質の立体構造情報の入手にはまだまだ未解決な問題が山積している。最も大きな問題は、その解析スピードとコストであろう。従って、わが国ではわずかな大手製薬企業だけしか着手できていない。そこで、本答申で述べられているような放射光施設の共同利用、あるいは蛋白質発現や結晶化における新技術の開発によって、構造生物学は創薬領域へさらに大きく寄与できる可能性を秘めている。
5.5.3.工業への応用
蛋白質の工業への応用は、将来的には全く新たな人工的機能の付加等も考えられるが、現在のところは、既に分子機能が既知の蛋白質を、その立体構造を基に改変して、工業的により有効なものにしようとする作業が現実的である。実際、その特異的分子認識の特徴を生かして、検査用試薬として多くの酵素蛋白質が利用されているが、工業的には、さらに高活性なものが望まれたり、安定性の高いものが望まれることが多い。
酵素反応等の活性を上昇させるのは、その反応メカニズムが推定されていても容易ではないが、基質や基質アナログとの複合体立体構造が既知であれば、その構造をもとに、活性を上げたり、より排他的な分子認識を目指すアミノ酸改変の候補を示唆できる。最終的にスクリーニングによって最適なものを探すことになっても、どの部位のアミノ酸に対して変異をかけるべきかは、やはり立体構造を基に考えることが効率的である。
また、蛋白質の安定性に関する研究はこの10年間に大いに進展し、日本人研究者の寄与も大きいが、その成果によって、どのようなメカニズムによって安定化がはかられるかの知見がまとまってきた。改変したい蛋白質の立体構造が既知であれば、ある一つだけの安定性向上設計を示すことは未だに困難ではあるが、いくつかの安定性向上のメカニズムを考え提示することは可能な状況になってきた。実際に、このようにして蛋白質の構造が安定化された多くの成功例が報告されている。熱安定化が達成された酵素の場合には、反応を効率が高くなる高温で行うことも可能なため、同時に高活性化という目的も果たせる場合がある。
一方、天然の蛋白質では、機能を果たす活性部位は極めて局所的であるにも関わらず、進化の歴史によるのか、その活性には必ずしも必要ではないと思われる他のドメインを携え、巨大な蛋白質となっている場合も多い。あるいは、別の活性を持つドメインが付随している場合もある。このような場合、立体構造を基に、ある特定の機能のみを発現するドメインだけを切り出してきたり、リンカーで必要な部位のみをつないだりする技術(蛋白質のミニマイゼーション)が有効である。例えばミニシャペロニンやミニインテインなどは、蛋白質のin
vitro合成におけるフォールディングやペプチド鎖ライゲーション等のバイオテクノロジー技術にたいへん有効に働くものと期待される。
![]()
6.わが国の研究・教育体制
6.1.研究体制の現状と問題点
6.1.1.大学などにおける研究体制の現状
我が国の主要大学には、理学部、薬学部を中心に、構造生物学を主要研究手段あるいは研究分野とする研究室がある。
全国で45程度の研究グループが、X線結晶構造解析を主要分野としている。大学における研究グループの規模は平均して小さく、スタッフが2名以下の研究グループ(教授と助手または助教授と助手、あるいは一人だけ)が半数を占める。スタッフ4名以上(技官を含める)を持つ研究グループは、全体の20%に満たない。それにもかかわらず、精力的に研究を展開しているグループが多い。研究グループの規模によるが、クローニングから蛋白質精製、結晶化、測定、構造解析までの全てを自前で行っているグループは少ない。多くの場合は、共同研究により、精製蛋白質の提供を受けるか、あるいは大量発現系の提供を受けるなどで、結晶化あるいは蛋白質精製以降のステップを行っている場合が多い。理化学研究所など大学以外の研究機関では、大きな規模の研究グループが複数存在している。このようなグループでは、生物学的研究対象を決め、蛋白質の同定、クローニングから蛋白質精製、結晶化、測定、構造解析までの全てを自前で行っている。
主たる研究課題は、以下のようなものである。情報伝達に関わる蛋白質群の構造解析、遺伝子発現調節に関わる蛋白質群の構造解析、エネルギー変換に関わる蛋白質の構造解析、酵素一般の構造解析、免疫他生体防御系の構造解析、超分子複合体(ウィル
ス、リボソーム、プロテアソームなど)の構造解析、方法論開発。
NMRを主要研究手段としている研究グループは、40程度である。比較的小さい規模の研究室が多い。NMRによる溶液構造解析を主たる研究テーマとしているグループは必ずしも多くない。溶液構造解析はNMRの能力のごく一部であることを反映していると思われる。ダイナミクスや蛋白質折り畳みなど蛋白質物性を研究しているグループも多い。
主たる研究テーマは、転写、情報伝達、翻訳など生物学的に重要な現象の理解を念頭においた立体構造解析、Structural
Genomicsに代表されるより多くの立体構造モチーフの解明、折畳みや動力学などの蛋白質の物性的研究、方法論の開発、固体NMRによる膜蛋白質の構造解析及び手法開発などである。
X線結晶構造解析グループとあわせ、日本の拠点的大学の中で、東北大学、名古屋大学および九州大学の寄与が少ないことが懸念される。
電子顕微鏡を主たる研究手段として用いている研究室研究グループは、医学部解剖学教室をはじめとして多数あるが、生体高分子の立体構造解析を主眼としているグループはそれほど多くはない。精力的な研究を展開しているのは、6グループ程度である。研究グループの規模は概して小さく、その中で装置の維持管理、研究を行わなければならないという研究環境の中で、精力的に研究を展開している。電子顕微鏡、電子線回折が汎用の立体構造解析手段となることは考えにくいが、世界のトップレベルにある研究グループのアクティビティーを継続させていかなければならない。そのための人材育成についても考える必要があるだろう。主たる研究テーマは、膜蛋白質の立体構造解析及びモーター蛋白質群の構造解析である。
X線繊維回折、溶液散乱を中心的研究分野としている研究グループは、20に満たない。研究グループの規模は概ね小さくほとんどが一人ないし数人の研究グループになっている。研究グループの規模を反映して、研究テーマにも片寄りが見られ、オリジナリティーもあまり高くないように思われる。その中で、蛋白質折畳み、動力学、モーター蛋白質群の構造解析などで世界のトップクラスにある3研究グループが際立った貢献をしている。中性子に関しては、日本原子力研究所のグループが唯一といってよい。
光学顕微鏡などにより、低分解能ながら一分子の細胞内での挙動を観測するという技術は、我が国オリジナルなものであり、構造生物学の重要な一端を担っている。この分野に関係する研究グループは、20程度あり、それぞれに精力的な研究を展開している。研究グループの規模は、標準的なところが多いように思われる。主たる研究テーマは、生体運動系、遺伝情報発現系、エネルギー変換系、膜蛋白質動力学など多岐にわたっている。一分子生理学、一分子生化学など新しい分野を切り開いていくことは間違いない。
構造生物学における理論の重要性は、立体構造及び配列のデータベースが充実すればするほど、増していく。シミュレーションを含み、理論を行っている研究グループは30程度である。研究グループの規模は概ね小さい。しかし、精力的に研究を展開しているグループが多い。オリジナルな構造解析支援ソフトウェアを開発してきたグループもあるが、ユーザーに恵まれず、米国のソフトに席巻されているのが現状である。研究能力は高いのであるから、実験サイドから積極的に支援する体勢が望まれる。研究テーマは、シミュレーション、分子レベルの博物学、デザイン、予測など多岐にわたっている。
6.1.2.共同利用研究施設の現状と問題点
6.1.2.1.SPring-8
SPring-8は我が国が世界に誇る第三世代放射光実験施設である。周長1.4kmを超える電子貯蔵リングに8GeVの光速に近い電子を細い電子束にして走らせ、最高100mAの電流としアンジュレータなどの挿入光源から収束性が高い高輝度X線ビームを産み出し、生物学や物性物理学の計測実験に利用する。構造生物学用ビームラインは現在8本、うちタンパク質結晶解析用に使われるのが7本、また何本かは溶液散乱、繊維回折、分光に使われる。理化学研究所や大阪大学蛋白質研究所の専用ビームラインを除く完全な共用ビームラインは2本である。
SPring-8キャンパスには、理化学研究所(理研)構造生物学研究系と高輝度光科学研究センター(JASRI)実験部門グループがあるが、異なる研究棟に所在し、研究上の連携は余り強いとは言えない。理研には5つの構造生物学関連研究室と、それに加えて放射光利用連携プロジェクトが2領域(各3、計6グループ)ある。このように構造生物学の一大拠点ではあるが、利用連携プロジェクトの1領域がやや共同利用的な性格を持つのと、理研ビームラインが部分的に他研究機関グループとの共同研究に利用され共同利用的性格を持つのみで、共同利用研究施設ではない。とはいえ、まだ部分的に実験棟建設中ということもあり、独自の研究活動が非常に高いレベルにあるとは言えない。JASRIはSPring-8という共同利用施設の運営管理機関であるが、独自の研究活動も推進する計画を持ち、実験部門を置いている。ただし、SPring-8が稼働し共用を開始してまだ2年半、この間ビームライン建設業務最優先のため利用促進部門共用ビームラインを支援するグループの研究員は増強されたが、実験部門の研究員はまだ少数で、ようやくその拡充計画段階にある。ビームラインを支援するグループの研究員は、いまだ日常の大半をビームライン建設とユーザー対応に追われており、独自の研究を進める余地は現在のところ非常に少ない。しかし、ビームライン建設が一段落しつつあるため、近い将来実験部門とビームライン支援グループの組織を改編して、JASRI独自の研究活動を開始し推進する計画のようである。
JASRIの問題は、利用促進部門が共同利用研究者の利用支援に追われており、インハウススタッフと共同利用研究者との間で研究上の連携が現時点では行なわれていない点と、独自の実験部門研究グループの拡充計画がまだよく見えない点である。その点では、つくばのフォトンファクトリーと同様の問題を抱えている。ただし、JASRIが本格的に独自の研究をと考えているのであれば、世界に誇る高輝度X線を最大限に利用できる立場にあるだけに、高いレベルの研究活動に発展するポテンシャルは高く、将来が楽しみである。理研の問題もそれに近いところがあり、主たるメンバーがビームラインの建設と共同研究の対応に大半の時間を費やしてきたため、独自の研究活動はいよいよこれからという印象が強い。両者ともに組織的な問題として、重要な生物学的問題に取り組むための体制と基盤をもったグループ形成ができていない点が指摘される。つまりそういった問題に取り組むために必須な、タンパク質の大量発現系構築(遺伝子工学)、蛋白質大量精製(生化学)、そして結晶化を行う人材を、有機的に配置するグループづくりにまで至っていない。JASRIおよび理研が、その豊富な人的および予算的資源を有効利用すれば決して不可能なことではなく、大所高所から先を見通す能力のあるリーダーの存在が重要な鍵となるであろう。
6.1.2.2.物質構造科学研究所(放射光実験施設)の現状と問題点
物質構造科学研究所においては、これまで坂部知平教授を中心に、イメージングプレートを検出器としたワイセンベルグカメラ(坂部カメラ)の開発と結晶構造解析の共同利用が精力的に行われてきた。坂部カメラは、放射光の利点を生かしたカメラとして、放射光利用研究になくてはならないものとなってきた。坂部教授の退官後も、坂部教授の指導のもとに、優秀な助手3名が共同利用を支援するとともに、装置の開発に携わってきている。坂部カメラ改良、時分割ラウエ法の開発にあたってハード、ソフト両面で支えてきた二人の助手がそれぞれ大学へ昇任転出し、物質構造科学研究所における蛋白質結晶構造解析グループのあり方が議論されてきた。その結果、大きな構造生物学研究グループを形成し、日本におけるセンターとして機能する必要があるとの観点から、教授候補者が選ばれ、候補者を中心にグループの再構築が行われることになっている。
繊維回折、小角散乱においては、検出器、光学系の専門家であったスタッフが転出した後に、構造生物学分野から助手が赴任している。物質構造科学研究所の共同利用におけるこの分野のアクティビティーの高さから、一層の充実が望まれている。幸い、選ばれた教授候補者は、小角散乱の経験を有しているため、広い視野からのグループの構築がなされると期待されている。
研究所の所内スタッフの研究の潜在能力は高いのであるが、ビームラインの技術支援スタッフの数があまりにも少ないことから、共同利用の支援にその時間の大半を取られ、スタッフ固有の研究時間がまとまって取れないという問題点がある。結晶構造解析グループでは、主だったユーザーグループから、共同利用支援にあたる学生の派遣を行っている。小角散乱グループでは、協力ビームライン計画により、実際の運営は、ユーザーグループで行い、所内スタッフのロードをできるだけ軽減することを考えている。しかし、マシンの運転期間中は、様々な形で、ビームラインの諸業務に時間が取られてしまう現状がある。スタッフの優先ビームタイム制度はあるが、それを支えるための試料調整にあたる時間、設備ともに貧弱であり、またデータ収集後解析に関わる時間も取れず、スタッフオリジナルな研究を推進することができないのが現状であった。さらに、放射光実験施設におけるスタッフには、新しい測定装置の開発、建設という業務も科せられる。坂部教授以下転出したスタッフは、装置開発の優れた手腕を有していたため、放射光結晶構造解析におけるPFの優位性を長い期間にわたって保持することができたのは忘れてはならない事実である。今後は、構造生物学そのものの研究と、装置の開発建設、さらに共同利用の環境整備という3本の柱をバランスよくこなしていくことが、各スタッフに要求される能力となろう。また、機構内の筑波大学粒子線医学研究センターの跡地を構造生物学研究棟に転換する計画が実現し、構造生物学用の研究室が完備されることになった。機構としては、構造生物学は今後の放射光科学を支える重要な一分野と認識しており、そのために様々な措置がなされてきている。優れたスタッフを得ることにより、物質構造科学研究所が構造生物学研究の一拠点として機能してくことは、疑いのないことである。
結晶構造解析の共同利用においては、結晶ができたときにすぐ測定ができるというシステムになっていない。前もって申請書を出し、採択されていなければならない。申請課題は2年間有効であるが、原則的に申請課題以外の実験を割り当てビームタイムで行うことはできない。テスト実験についても原則的にP型課題として申請する必要がある。結晶構造解析に関して言えば、ビームタイムアサインにはもう少し融通が利いてもいいのではないかと考える。そのためのシステムを整備する必要があろう。
6.1.2.3.NMR共同利用の現状と問題点
ゲノム科学総合研究センター(GSC)における、高磁場NMR装置群はその規模において、従来の欧米の研究施設の常識を遙かに越える規模のものであり、いわゆる「NMR
Park」として国際的にも大きな波紋を呼んだ。しかしながら、この施設は(少なくとも現時点では)共同利用研究施設としての機能を想定して運営されるものではなく、NMRによる蛋白質の立体構造研究の効率化を追求するための構造解析施設として捉えられるべきものであろう。欧米においては、理研の主導する本プロジェクトの立ち上げを契機として、類似した高磁場NMRの共同利用研究施設の構想が次々と策定され、実際に予算がつきつつある。これらの研究施設にはポストゲノム研究や蛋白質立体構造決定を中心的課題として掲げているものも含まれるが、基本的には生体高分子研究の推進に向けた高磁場NMR共同利用研究施設を各地に形成することを目標としている。
蛋白質の立体構造を効率良く決定する技術としては、X線解析法が優位にあるという考えは、欧米においては広く認められている。このような構造決定手段としてのNMR技術の限界に関しては、欧米諸国ではいわば共通認識化しているにも関わらず、幾つもの高磁場NMR装置を中心とする共同研究施設が設立されつつある点に注目すべきであろう。このことは、分子量限界を初めとするNMR技術の構造決定手段として限界が、生体高分子の構造研究手段としてのNMR技術の総合的価値をいささかも下げるものではない、との考えが根底にある。我が国の生体高分子に関するNMR研究推進の重要性は明白である。と同時に、NMR研究の推進は大型装置の集積や、研究者の一極集中化により達成されるものではないことも同様に明白である。我が国が基礎研究の立ち後れと、独創性ある研究が乏しい現実は、確かに我々にとって苛立たしくも深刻な事態である。欧米と比肩して勝るとも劣らぬ装置大国となった現在も、世界をリードする独創的技術を発信できないのは何故であろうか。このような、いわば逆説的には、我が国の伝統的ともいえる問題を根本的に解決するために、果たして研究体制を論議し改変することで十分かどうかと言う点が気がかりである。
客観的に眺めるならば、我が国においても、世界をリードする分野こそ数少ないものの、それなりの成果と実力を持つNMR研究グループがある。これらのグループは、個々には不満はあるにせよ、かつての状況とは比べることもできないほどの潤沢な研究費と大型装置を持っている。それらのグループはそのリーダーによりそれぞれ異なった分野を得意としており、このような異質な研究室が互いに切磋琢磨することにより、時間は掛かるが、初めて世界に抜きんでた研究グループが誕生するのではないかと思う。時間はかかるが、正当な研究成果への評価と研究助成が機能するならば、一番真っ当な基礎研究推進の王道であろう。むしろ、多くのグループでは研究費の継続性や、装置の更新に過剰な時間をとられたり、或いは研究員や補助職員の確保など、今までに言い古された問題点に悩んでいるのではなかろうか? これらの軽減は確かに有効な推進手段である。NMR装置はなかなか気むずかしいところがあり、じっくりと時間を掛けて使い込まなくては良いデータを与えて呉れない面がある。共同利用施設であれば、高い専門性を持つ技術者や研究者が、外部からの研究者の要望に対応した実験を、高レベルで実施して貰えるのでなければ独創的な研究を生み出す役にはたたない。かといって、数少ない人材を大学や研究所で奪い合い、人材を育成する手段を持たなければ早晩基礎研究は破綻するだろう。
共同利用NMR研究施設が大きなインパクトを与えるためには、大型装置を利用できる機会の提供だけではこのように設備が行き渡った現在では不十分である。この点は高輝度X線源の利用とは根本的に異なる。むしろ、研究者が期待するのは、自らのグループでは到達できない高い水準での測定、解析、さらには試料調製技術である。高度な固体NMR測定、高圧NMR測定等の特殊測定技術は頻度は少なくとも必要な技術であろう。高度な安定同位体標識技術の利用も、この範疇に入る。これらはそれぞれ専門性の高い研究グループを集中して研究施設に集めることも可能ではあろうが、それらの研究が行われている研究室を一種のCOEとして認定し、補助、育成し、その代わりに他の研究者が技術にアクセスできるシステムにすることも一考の余地がある。特に研究者の育成や学生の教育には、大学のシステムを利用できるだけに優れた面がある。グループリーダーの停年の問題もここには関連してくるが、米国にならって全て研究成果の優劣により判断し、他の差別要素を排除する考えも取り込まなければなるまい。これらの独自技術を十分に評価せず、絶やすようなことがあれば、我が国で今後様々な独創的技術を育成することは望めまい。
時に応じて学問の呼び名が変わることがあっても、従来も、現在も、そして将来にわたっても蛋白質や核酸等の生体高分子の構造研究に携わる研究者は等しく、それらの持つ多彩な生物機能を立体構造に基づいて説明することを究極の目的としてきたのではあるまいか。構造生物学という言葉が人口に膾炙される以前より、多くの研究者がNMR、X線、電子顕微鏡に惹きつけられてきたのは、全ての生物機能は分子構造とその時間的変化により説明できる筈であると信じていたからであろう。ゲノム科学、構造生物学、ポストゲノム、プロテオーム等を定義し、新たな学問領域であると主張するのはよいが、スローガン倒れにならぬよう、これらの分野の多くの研究者の知恵と意見を生かす体制を整えなければならない。
6.1.2.4.データベースの現状と問題点
データベース管理の基本的な体制としては、データベースを恒常的に維持できる組織が必要である。期限つきのプロジェクトでは、たとえ10年程度の長いものでも、プロジェクト終了と同時にデータベースを終了させるわけにはいかない。このデータベースの特殊性を理解し、国家の資産として維持・管理する体制が、基本的に必要であり、今後の構造生物学の展開に対応するためには情報インフラストラクチャーの整備が不可欠である。
構造生物学データベースは、その内容の専門性と利用方法の特殊性のため、データベースの設計と公開・利用のしかたに関しては、構造生物学の専門家・研究者が方針を立てる必要があり、また日常的なデータ管理に関しても構造生物学の専門的視点からのデータ編集、入力等が必要である。一方、データベース運用、プログラミング等を円滑に行うため、コンピュータ技術者も必須である。これらの人材を育成したり、雇用を確保する努力が欠けているのが現状である。
最近の科学予算は大型化してはいるが、プロジェクト指向が強く、一定の期間で終了することを前提としているものが多い。現在、運営・維持されているデータベースの多くが、これらのプロジェクト予算に依存している。一方、データベースは、ある時点でデータがなくなって終了するというものではなく、世界中に利用者がいるかぎりは維持・運営する国際的な義務が生じる。また、データが加速度的に増加するため、年ごとに大型化し、その維持に必要とされるコンピュータ経費も増加せざるをえない。また、上記した人材確保のための人件費も、現状では、単に金額が不足しているだけでなく、その支払を行う予算項目すらないことも多い。さらに、データベース運営のための国際協力が広がっており、そのための海外出張費も必要とされる。このようなデータベースの特殊性を理解し、データベースのための予算を、その総額と利用しやすさとを考慮し、長期的な視点で確保していく必要がある。
構造生物学データベースを維持・管理する生物学の専門家・研究者は、必ずしも情報科学の専門家と交流しているわけではなく、技術のニーズがうまく伝わっていない。データベースのフォーマットやデータの標準化の決定にも、情報科学の専門家のアドバイスは重要とされるが、必ずしも交流は盛んでない。そのため、市販の高価なリレーショナル・データベースを購入せざるをえないことも多い。さらに、データベース管理のための人材の教育についても、情報科学の専門家との密接な協力体制が必要とされる。一方、情報科学の専門家が構造生物学データベースに関わる様々な研究を遂行していくためには、構造生物学、少なくとも生物学についての素養が求められる。こうした観点からも、構造生物学研究者と情報科学研究者の積極的な交流が望まれる。
データベースの重要性は、最近認められつつあるが、その構築・維持・管理を行っている構造生物学研究者の業績に対する研究者集団からの評価は、依然として高くない。特に、大学の教官に対する業績評価は、原著論文を中心としてなされているため、現状では、研究を別途に行って論文を発表しながら、データベースも運営するという状況が続いている。優秀な人材によって、質の高いデータベースを構築・維持・管理していくためにも、データベース構築・運営に対する一般社会および研究者社会の評価を高めていく必要がある。
6.2.人材育成体制
上記で見たように、大学での構造生物学の教育研究体制は必ずしも十分ではない。大学における研究グループの規模の小ささを反映している。結晶構造解析やNMR溶液構造解析の技術の進歩は著しく、結晶構造解析に関していえば、ルーチンとしての構造解析は確立しており、人さえいれば約1年で解けるようになった。また、NMRにおいても、培養から構造解析まで3ヶ月を集中して学べばこなせるようになっている。さらに、最近の研究費のつき方が、プロジェクト研究志向になっているため、大学院生もプロジェクトの一員として組み込まれるようになってきている。このような中で、構造解析技術の原理・基礎からしっかりと教育していくことには困難が生じている。この結果、新しい方法論の開発に取り組むことのできる人材の供給に問題が生じている。構造ゲノム科学などのプロジェクトにより、短い期間にたくさんの構造を解くことが求められている。そのためには、今の方法論のスケールアップではとうてい追いつかず、全く新しい構造解析法、試料作成法などの確立が急務である。一通りの構造解析のできる人材、方法論の開発までできる人材それぞれが必要とされている状況であるが、両者の育成が遅れているのは、ゆゆしい問題といえる。プロジェクトの展開によりポスドクのポジションは増えているが、ポスドクの立場から見ると、その後どうなるか不明であるという不安定さがある。
一方、構造生物学研究グループの数もあまり多くなく、研究グループの規模も比較的小さいことから、正常な競争原理が働きにくいという状況がある。研究室内での競争や批判が正常な運営に必要であるが、研究室間の人の流れは滞りがちである。これは構造生物学だけの問題ではなく、日本の科学研究全般にわたる問題でもある。
![]()
7.提言 構造生物学推進のあるべき姿
7.1.構造生物学研究の3本柱
現時点では構造生物学研究の様々なフロンティアは、次の3本の柱にまとめて考えることができる。これら3本の柱のいずれをも注意深く推進していかなければならない。
1)重要な生物学上の問題を構造に基づいて解明する研究
2)特定の蛋白質集団の蛋白質の構造を網羅的に調べ上げる研究
3)最先端の技術・方法を構築する研究
上記1)、2)の研究の展開においても、現状では3)は不可欠でこの面への配慮が特に大切である。
7.2.多様性なサイズ・性格の研究室の全国的配置
構造生物学は放射光等の大型施設を用いることを除けば、本質的にはスモールサイエンスである。さらに、対象・方法の多様性にその特徴がある。各研究者のキャリアパスを考慮に入れ、国内の構造生物学研究全体の活性を高く保つためには、研究室のサイズにも多様性を持たせ、最初は小研究室で業績を挙げ、その中の優れたリーダーが大研究室を組織する流れを国全体として作るべきである。
構造の実験的決定に携わる研究室において必要とされる能力・作業には、次のような面がある。
a)クローニング等の分子生物学的操作
b)蛋白質科学的操作
c)構造解析
d)装置の維持・管理
クローニングから構造解析まで一貫して自前でできるのが、理想的な研究室のスタイルであろう。そのための標準モデルとして、グループリーダー1名、博士研究員5〜6名、リサーチアシスタント・大学院生4〜6名と短期滞在の外国人研究者枠1〜2名のほか秘書業務等のサポート体制からなる研究室が考えられる。これ以上大きい研究室はむしろ効率が良くない。しかし、総ての研究室が上記のサイズを持つ必要はない。最低5人程度からなるグループでも、共同研究施設の利用、他の研究室との協力関係のあり方を工夫すれば、十分成果を挙げることができる。小研究室は総ての作業を自前でする必要はない。サイズの多様性が、研究者の流れを良くし、適度の競争を促し、全体としての活性化につながる。
7.3.理論的研究の必要性
構造生物学における構造決定は手段であって目的ではない。得られた構造から生物学的に重要な知見を得るには、しばしば深い理論的考察を必要とする。従って、理論研究グループは構造生物学研究の重要な構成要素である。主要な研究・教育の拠点には、実験研究室と共に理論研究室を配置しなければならない。理論研究室は、常に実験研究室と密接な交流・協力関係を維持していなければならない。
7.4.全国的研究体制
7.2で述べた全国的配置の具体像として次の3つのレベルの研究組織を整備すべきである。
7.4.1.拠点研究複合
全国5、6箇所程度の拠点に研究複合を設ける。その候補としては、(1)横浜市の理化学研究所ゲノム科学センターと横浜市立大学の研究室、(2)播磨の理化学研究所研究室・JASRI・姫路工業大学の研究室、(3)つくばの物質構造科学研究所の構造生物学研究室、(4)大阪大学蛋白質研究所の構造生物学関係の研究室、(5)遺伝学研究所の構造生物学関係の研究室、(6)日本原子力研究所の構造生物学関係の研究室、等が考えられる。これらの拠点に複数の構造解析に携わる研究室と理論研究室からなる研究複合を形成する。現在大阪大学蛋白質研究所が関与している立体構造データベース構築の活動は、格段の充実を図り世界的拠点の一つとしての役割を担わなければならない。
蛋白工学研究所およびその後身の生物分子工学研究所において達成された高いレベルの構造生物学研究の実績は、是非とも引き継がれなければならない。
上記候補の中で、科学技術庁系の研究施設が目立つ。それは人事・研究費配分の柔軟性がもたらした点が無視できない。一方で費用効率の面で懸念がある。高い研究レベルを維持し、先端的研究を推進するためには、これら研究施設における研究全般を研究者社会に格段に公開し、活発な議論を促す必要がある。
一方文部省系の研究施設は、研究のレベルは高いが、激しく変化する状況へ適応できる能力と環境を著しく欠いている。このことを自覚し、改善への格段の努力が必要である。
7.4.2.拠点大学に於ける研究・教育複合
全国5〜10箇所程度の主要大学に幾つかの研究室からなる研究・教育複合を設立すべきである。過渡的には、大学内に存在する複数の構造生物学関係の研究室が部局の枠を越えて密接に協力して研究・教育に携われる十分な予算的措置を伴った体制を作るべきである。これら拠点大学における研究者育成のための教育面での機能は特に大切である。
7.4.3.幅広い展開
上記以外に全国に100程度の研究室が展開していることが必要である。特に、大学の医学、生物学、農学、薬学等の学部・研究科に構造生物学に携わる研究室を必ず設置することは、構造生物学にとってもわが国のライフサイエンス全体にとっても切実に求められている。
7.5.構造生物学の総合的展開へ向けた研究者社会の対応
構造生物学は多様な方向に展開しつつ、新たな生命科学の中核を形成しつつある。用いられる方法も多様で、しかも激しく発展しつつある。多様な方向は総て純粋な基礎と応用との間の距離が短い。この短さ故、国際的な知的財産権を巡る競争に立ち向かわざるを得ない。この状況の中で、研究のための財政的・物的資源を適切に配分し、人的資源を急速に育成しつつ適切に配分しなければならない。この複雑な問題に研究者社会は叡智を持って立ち向かわなければならない。
![]()
Copyright 2002 SCIENCE COUNCIL OF JAPAN